Ventricular Septal Defects
Stephanie Fuller
VENTRICULAR SEPTAL DEFECTS
Ventricular septal defects (VSDs) are among the most common congenital heart anomalies. Isolated VSDs represent approximately 20% to 30% of all congenital cardiac malformations and have a prevalence of 1 to 2 per 1,000 live births. There is a slight female predominance.
Roger first described the clinical signs and symptoms of a VSD in 1879 as a small, flow-limiting VSD with associated normal pulmonary artery. At the other end of the clinical spectrum is a large VSD with severe pulmonary hypertension and fixed pulmonary vascular resistance resulting in cyanosis with right-to-left shunting through the VSD, first described by Eisenmenger in 1897.
VSDs may be associated with a wide variety of other cardiac defects, including mitral valve disease, atrioventricular (AV) discordance, conotruncal abnormalities such as transposition or double-outlet ventricle, and hypoplasia of either ventricle. They are the most common heart defect in many chromosomal anomalies, including patients with trisomies 13, 18, 21 and with Holt-Oram syndrome. Isolated VSDs and those associated with patent ductus arteriosus and coarctation of the aorta are described in this chapter.
CLASSIFICATION OF VENTRICULAR SEPTAL DEFECTS
The ventricular septum forms between 4 and 7 weeks of gestation and involves fusion of several tissues, including endocardial cushion-derived mesenchyme, primary atrial septum, and muscular components of the atrial and ventricular septum. The muscular intraventricular septum forms by infolding of the ventricular muscle within the primitive cardiac tube. It then aligns with the conal septum that is positioned between the two outflow tracts. The membranous septum closes adjacent to the anteroseptal commissure of the tricuspid valve. VSDs may occur anywhere in the septum.
VSDs are classified based upon their location within the ventricular septum. Spontaneous closure rate and the presence of associated defects depend upon the location of the defect and the type of VSD. Of the many classifications of VSDs, the most widely accepted today are those of Soto and van Praagh (Fig. 80.1). The ventricular septum has three main components: (1) an inlet portion beneath the septal leaflet of the tricuspid valve and extending from the tricuspid annulus to the papillary attachments of the tricuspid valve chordae, (2) a trabecular or muscular portion that extends from the chordal attachments of the tricuspid valve to the apex of the ventricles and cephalad to the conal septum, and (3) a smooth-walled conal or outlet septum, which comprises the infundibular septum clasped between the anterior and posterior limbs of the septal band (trabecular septomarginalis) and extends up to the pulmonary and aortic annuli. The inlet and trabecular portions are often referred to collectively as the ventricular septum, in contrast to the conal septum, which is also called the “outlet” or “infundibular” septum.
Conoventricular (Perimembranous) Defects
Conoventricular defects are the most common isolated VSDs, comprising about 70% to 80% in most series. They are situated in the junctional area between the conal or outlet and inlet portions of the septum and may extend into the inlet, outlet, or both parts of the septum. They extend up to the tricuspid valve annulus in the region of the anteroseptal commissure of the tricuspid valve. The aortic valve is easily visible through the defect when viewed through the tricuspid valve at operation. Alternatively, these defects may be partially occluded or rendered restrictive by overlapping tricuspid valve tissue. Occasionally, the noncoronary aortic leaflet may prolapse through the defect resulting in progressive aortic incompetence.
The conduction system is intimately associated with this VSD. The bundle of His penetrates the right trigone of the central fibrous body at the anteroseptal commissure of the tricuspid valve. From there, it is closely related to the lower half (posteroinferior edge) of the defect, giving off the left fascicles along its course to the medial papillary muscle (also called the muscle of Lancisi or the papillary muscle of the conus). As it passes just inferior to the latter, only the right bundle remains, and this continues into the trabecular septomarginalis away from the edge of the defect. Conoventricular septal defects are usually repaired through the right atrium.
Conal Septal Hypoplasia (Outlet) or Conotruncal Defects
Conotruncal defects are also called supracristal, subarterial, subpulmonary, juxtaarterial, or infundibular. They account for 5% to 10% of isolated defects, except in the Asian population in whom they are more common accounting for up to 30% of VSDs. Typically, the defects are oval in shape and extend up to the pulmonary and aortic annuli. Because the normal subpulmonary conus is deficient, the pulmonary and aortic valves are separated only by a very thin rim of fibrous tissue that maintains fibrous continuity between the aortic and pulmonary valves. This results in aortic leaflet prolapse, which occurs in 40% to 50% of conal defects. The right aortic leaflet most commonly is sucked into the defect, resulting in aortic insufficiency. The conduction tissue is remote from the borders of this type of VSD, which is most easily repaired through a short transverse incision in the right ventricular outflow tract, or through the main pulmonary artery (Fig. 80.1). These defects will not close spontaneously
but can be significantly restricted by a prolapsed aortic valve leaflet.
but can be significantly restricted by a prolapsed aortic valve leaflet.
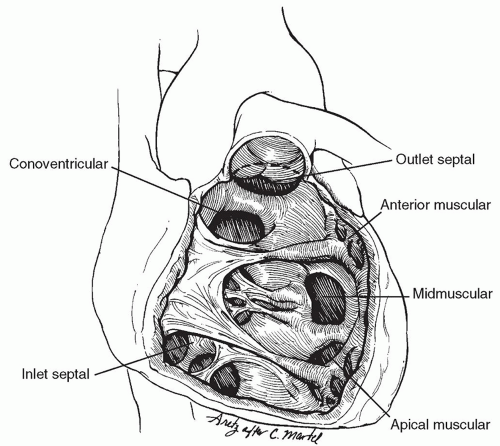 Fig. 80.1. The right ventricular free wall has been resected to show the ventricular septal defects: conoventricular (perimembranous); conal septal (subpulmonary or outlet); atrioventricular (inlet); muscular that may be midmuscular, anterior, or apical. The penetrating bundle is closely related to the inferior margin of the conoventricular defect and diverges away from this margin into the trabecular septomarginalis beneath the muscle of Lancisi. |
Inlet Septal Defects (Atrioventricular Canal-Type Defects)
Inlet septal defects account for about 5% of isolated defects. They are situated beneath the septal leaflet of the tricuspid valve, with the tricuspid valve annulus forming their posterior border. The tricuspid valve may straddle across the defect with attachments into the left ventricle. The conduction tissue is closely related to the posteroinferior border of the defect, up to the muscle of Lancisi, at which point the right bundle branch diverges down the trabecular septomarginalis to form the moderator band. The AV septum is intact, although occasionally the anterior leaflet of the mitral valve may be partially cleft. Inlet septal defects will not close spontaneously and are best repaired through the tricuspid valve.
Muscular Defects (Trabecular Defects)
Muscular VSDs account for 10% to 20% of isolated VSDs, may be single or multiple, and may occur in any part of the muscular septum. They may be associated with other types of VSDs. They are generally classified depending upon their location in the septum and may be divided into midmuscular defects (most common), apical or posterior muscular, and anterior muscular defects (Fig. 80.1). Muscular defects may have numerous openings of variable size on the right ventricular side but only a single opening on the left ventricular side of the septum. This is referred to as a “Swiss cheese” defect. The conduction tissue is generally remote from the edges of a muscular defect, with two notable exceptions: (1) when associated with a conoventricular defect, the penetrating bundle usually runs in the muscle bridge separating the two defects and may easily be injured if each are closed individually and (2) if a muscular defect is present in the inlet septum (i.e., the defect is separated from the tricuspid valve by a thin rim of muscle), the conduction tissue runs along the defect superior and anterior (leftward) margin— the AV node penetrates the interventricular septum at the anteroseptal commissure of the tricuspid valve and takes the most direct route to the medial papillary muscle along the superior border of the defect. Muscular defects often close spontaneously. Surgical approach is variable based upon the location of the defect.
PATIENT CHARACTERISTICS
Both the relative pulmonary vascular resistance and the size of the defect determine the hemodynamic effect of the VSD. In the early newborn period, when pulmonary and systemic vascular resistances are relatively equal, there is little or no shunting of blood across the defect. As the pulmonary vascular resistance declines in the first weeks of life, the degree of shunting across the defect increases. Shunting occurs largely during systole and is directed to the lungs and the left side of the heart, causing left heart overload. In infants with a hemodynamically significant VSD, increased pulmonary blood flow from moderate-to-large defects leads to symptoms and signs of heart failure and to left atrial and left ventricular dilation.
Infants and children may not develop symptoms early in life unless the VSD is large. Patients with symptomatic VSDs present similar to those of heart failure, including tachypnea, tachycardia, increased work of breathing and, in some cases, repeated pulmonary infections. Those patients with large VSDs are often too tachypneic to feed orally, experience recurrent chest infections and aspiration of gastric contents, and may have pulmonary hyperinflation syndrome or cardiac asthma. The latter results from systemic pressure in the segmental pulmonary arteries compressing the small bronchi and resulting in chronic air trapping. Many of these infants fail maximal medical management aimed at decreasing the symptoms of heart failure.
DIAGNOSIS
The size, number, and location of VSDs can be accurately defined by echocardiography. Estimates of pulmonary artery pressure can be made using Doppler imaging of the velocity of a tricuspid regurgitant jet, if present. Defects are considered “large” if they approximate the size of the aortic annulus or result in systemic pulmonary artery pressures. Chronic elevation in
pulmonary blood flow can alter the pulmonary vascular bed, leading to intimal proliferation and muscularization of the arterioles known as Eisenmenger syndrome, a condition where the increased pulmonary vascular resistance leads to pulmonary hypertension and right-to-left shunting across the VSD. Ultimately, this can result in cyanosis.
pulmonary blood flow can alter the pulmonary vascular bed, leading to intimal proliferation and muscularization of the arterioles known as Eisenmenger syndrome, a condition where the increased pulmonary vascular resistance leads to pulmonary hypertension and right-to-left shunting across the VSD. Ultimately, this can result in cyanosis.
VSDs are also characterized by the degree of shunting by catheterization. A simple formula for calculating the Qp:Qs ratio using oxygen saturations measured by cardiac catheterization is as follows:
Qp:Qs = (Ao%−RA%)/PV%−PA%)
“Medium”-sized defects result in a Qp:Qs ratio of 2:1 to 3:1 and a systolic pulmonary artery pressure that is 40 to 50 mmHg or about one-half that of the aorta. “Small” defects have essentially normal pulmonary artery pressures and a Qp:Qs ratio of <1.5:1.
 INDICATIONS FOR OPERATION
INDICATIONS FOR OPERATIONMore than 75% of small VSDs will close by fibrosis and muscular hypertrophy within the first 2 years of life. Closure of a VSD is recommended once a large VSD is detected or once a patient becomes symptomatic. Thus, infants with large VSDs presenting in the first few months of life with severe congestive heart failure should undergo prompt repair. Delaying surgery until the patient is “bigger” is not beneficial and often results in additional morbidity and mortality.
Historically, small infants with a large VSD were initially palliated by placing a constricting band around the main pulmonary artery, suturing this to the adventitia, and gradually narrowing the band circumference until the systolic pressure in the pulmonary artery distal to the band was reduced by 50%. This decreased the flow through the pulmonary artery, ameliorated congestive cardiac failure, and allowed the patient to grow to a large enough size for the band to be removed and the VSD to be safely repaired. However, as methods of myocardial protection advanced and the surgical skills and postoperative care of small infants improved, pulmonary artery banding was abandoned in favor of primary closure of the VSD except in rare cases.
When reviewing the indications for surgical closure of a VSD, there are four aspects to take into consideration: (1) characteristics of the defect, (2) the patient’s age and symptoms, (3) pulmonary vascular resistance, and (4) associated cardiac and noncardiac defects.
Any VSD associated with failure to thrive should be closed. If a VSD is associated with prolapse of the aortic valve leaflet resulting in even mild regurgitation, this should be repaired irrespective of the symptoms or size of the defect. In addition, surgical repair is recommended for inlet or outlet types as they are not likely to close spontaneously. The same is true for residual VSDs typically >3 mm in size.
The exception to early primary closure in a symptomatic infant is the presence of multiple muscular defects, which is still associated with significant mortality when repaired in infancy. This is one of the few remaining indications for pulmonary artery banding. Debanding of the pulmonary artery and repair of the VSDs are done when the patient is about 6 to 12 months of age. Some muscular defects may be suitable for percutaneous catheter device closure by our cardiology colleagues. This can be done either before operation, intraoperatively with a hybrid approach or postoperatively. Apical and anterior muscular defects are often difficult to adequately close in the operating room and may be closed percutaneously.
Infants with symptoms controlled with medical therapy and large defects that have not appreciably decreased in size should be electively repaired between 6 months and 1 year of life. Most of these infants experience poor weight gain and failure to thrive. Many will develop increased pulmonary vascular resistance. These infants are considered to have “reactive pulmonary hypertension” and are at increased risk for developing pulmonary hypertensive crises in the postoperative period, particularly if their surgery is delayed.
Beyond 12 to 18 months of age, patients with large VSDs should undergo cardiac catheterization and be repaired if their pulmonary vascular resistance is <8 to 10 U/m2 while inspiring 100% oxygen or on nitric oxide thus demonstrating reversibility or the Qp:Qs ratio exceeds 1.5:1 at rest. Pulmonary vascular resistance is seldom prohibitive during the first year of life in patients with an isolated VSD, except occasionally in patients with associated Down’s syndrome. These patients tend to develop pulmonary vascular obstructive disease at an earlier age.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


