Aortopulmonary Window
Moritz C. Wyler von Ballmoos
Michael E. Barnes
James S. Tweddell
EPIDEMIOLOGY, EMBRYOLOGY, AND ANATOMY
Aortopulmonary window is a rare defect and accounts for about 0.1% to 0.2% of all structural congenital cardiac defects. Major academic centers could anticipate taking care of one or two infants with aortopulmonary window per year. Antenatal diagnosis of aortopulmonary window is challenging and delayed diagnosis may occur; the actual defect prevalence at birth is hence more difficult to estimate.
Aortopulmonary window is caused by failure of fusion of the two opposing conotruncal ridges that are responsible for separating the truncus arteriosus into the aorta and pulmonary artery. The aortopulmonary window, therefore, occurs between the two structures that normally result from septation of the truncus arteriosus, namely, the ascending aorta and the main pulmonary artery. Normal anatomy of the aortic and pulmonary valves separates this defect from the persistent truncus arteriosus. The similarity among conotruncal defects raises the question of a common underlying pathogenesis, but anatomic studies suggest that it develops by a different mechanism than truncus arteriosus. Furthermore, to date, no genetic association or environmental risk factors have been linked to aortopulmonary window.
A classification of aortopulmonary window based on the location has been proposed by Mori dividing aortopulmonary window into three types; proximal (type I), distal (type II), and total (type III). The classification introduced by the Society of Thoracic Surgeon adds a fourth, “intermediate” category accounting for the fact that this defect likely has a continuum of morphologies. For patients with aortopulmonary window without additional lesions, it is the size rather than the location of defect that impacts management and outcome (Fig. 81.1).
Origin of the right pulmonary artery from the ascending aorta and arch hypoplasia with interruption or coarctation are additional anomalies occurring with large aortopulmonary windows. With increasing size of the aortopulmonary window aberrations in flow result in abnormal incorporation of the right sixth arch, destined to become the right pulmonary artery, such that the right pulmonary artery arises from the rightward aspect of the ascending aorta. Similarly, with large aortopulmonary windows flow patterns can be disturbed such that there is preferential flow through the ductus arteriosus and diminished flow in the developing aortic arch resulting in distal arch hypoplasia including coarctation or interrupted aortic arch. It has been suggested that when associated with interrupted aortic arch, aortopulmonary windows are larger with greater distal extension. The Congenital Heart Surgeons’ Society multi-institutional study of aortopulmonary window with interrupted aortic arch found that all types of aortopulmonary window were more or less equally represented among patients with interrupted aortic arch (Fig. 81.2). Aortopulmonary window is not associated with DiGeorge syndrome, suggesting that aortopulmonary window is a distinct malformation not related to abnormalities of the conal septum, such as interrupted aortic arch with ventricular septal defect (VSD), tetralogy of Fallot, and persistent truncus arteriosus.
In reports based on the cumulative experience at high-volume centers, aortopulmonary window was associated with other defects in 58% of cases, the most common being ventricular and atrial septal defect, interrupted aortic arch or coarctation of the aorta, tetralogy of Fallot, and transposition of the great arteries. Abnormal origin of the coronary arteries is also commonly associated with aortopulmonary window. The coronary arteries may arise from the edge of the defect, or the origin may occur just on the pulmonary artery side of the defect.
PRESENTATION, DIAGNOSTIC CONSIDERATIONS, AND INDICATIONS FOR SURGERY
Critical to timely diagnosis and early repair, antenatal diagnosis of aortopulmonary window was first reported in 2002. Simple aortopulmonary window may not be identified by fetal echocardiography because equal pressure in the ascending aorta and pulmonary root in the fetus results in minimal detectable flow through the defect. Posterior deviation of the outflow septum that is characteristic of patients with interrupted aortic arch with a ventricular septal defect and would prompt further interrogation of the arch by the fetal echocardiographer, is lacking in the fetus with aortopulmonary window. The antenatal diagnosis of aortopulmonary window with interrupted aortic arch has only recently been reported.
The presentation of patients with aortopulmonary window may be different depending on the size of the window, associated other defects, and age at presentation. Although small, restrictive aortopulmonary windows do occur; generally, the communication is large, and patients present in the first weeks of life when pulmonary vascular resistance drops and increased pulmonary blood flow results in congestive heart failure. The clinical presentation is then similar to that of other patients with left-to-right shunts, such as patent ductus arteriosus (PDA) or VSD. This presentation includes signs of congestive heart failure such as tachypnea, diaphoresis, poor feeding, and inadequate weight gain. In early infancy, cyanosis is usually not a prominent feature, but with large defects, bidirectional shunting can produce systemic desaturation. With delayed diagnosis and persistent pulmonary overcirculation, remodeling of the pulmonary vasculature, and ultimately pulmonary hypertension occur.
Physical examination demonstrates a tachypneic infant with accessory respiratory
muscle use. Cardiac examination reveals an enlarged heart, and similar to patients with PDA, the pulses are bounding. A systolic murmur and accentuated second heart sound that is not split can be heard along the left sternal border; however, unlike the situation with patients with a PDA, a diastolic component to the murmur is rare. Chest X-ray films reveal cardiomegaly and increased pulmonary vascular markings consistent with increased pulmonary blood flow. Patients with associated arch abnormalities have pronounced left-to-right shunting and PDA-dependent perfusion of the lower body. With impending ductal closure there will be worsening pulmonary edema and shock.
muscle use. Cardiac examination reveals an enlarged heart, and similar to patients with PDA, the pulses are bounding. A systolic murmur and accentuated second heart sound that is not split can be heard along the left sternal border; however, unlike the situation with patients with a PDA, a diastolic component to the murmur is rare. Chest X-ray films reveal cardiomegaly and increased pulmonary vascular markings consistent with increased pulmonary blood flow. Patients with associated arch abnormalities have pronounced left-to-right shunting and PDA-dependent perfusion of the lower body. With impending ductal closure there will be worsening pulmonary edema and shock.
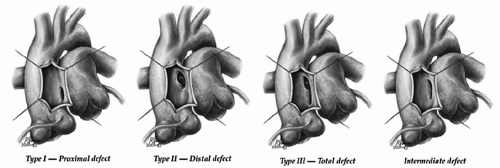 Fig. 81.1. The Society of Thoracic Surgeons’ classification of aortopulmonary window. (Reprinted from Backer CL, Mavroudis C. Surgical management of aortopulmonary window: a 40-year experience. Eur J Cardiothorac Surg 2002;21:773-779, 202, with permission from Elsevier.) |
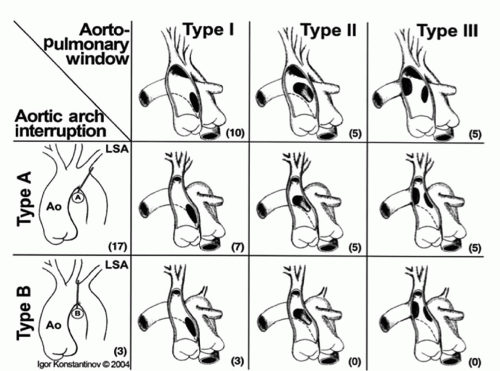 Fig. 81.2. The observed morphologies of aortopulmonary window with interrupted aortic arch. Type A interrupted arch predominated but the position of aortopulmonary window was more or less evenly divided between proximal and distal. (Reprinted from Konstantinov, IE, et al. Surgical management of aortopulmonary window associated with interrupted aortic arch: a Congenital Heart Surgeons Society study. J Thorac Cardiovasc Surg 2006;131:1136-1141, with permission from Elsevier.) |
The diagnosis is routinely made with 2D Doppler echocardiography. The location and size of the communication as well as associated anomalies are carefully identified. Echocardiography of the conotruncal region will identify the defect and associated shunt and the diagnosis of aortopulmonary window is confirmed when two separate semilunar valves can be identified. For surgical planning, echocardiographic examination should identify the coronary anatomy, the size and distal extent of the aortopulmonary defect as well as associated lesions such as anomalous origin of the right pulmonary artery and interrupted aortic arch. Echocardiography as the sole imaging modality has been shown to be accurate and sufficient for preoperative evaluation of even complex congenital defects including aortopulmonary windows with or without associated other defects.
Although using cardiac catheterization to assess the origin of the coronary arteries is theoretically appealing, the large defect occurring just above the sinuses of Valsalva combined with the tremendous pulmonary flow makes assessment of coronary artery anatomy with catheterization impractical. Cardiac catheterization should thus be reserved for the patient who presents after early infancy and therefore is at risk for elevated pulmonary vascular resistance.
Those patients who are found to have an elevated pulmonary vascular resistance should undergo testing with pulmonary vasodilators to determine whether the pulmonary vascular resistance can be reduced prior to surgical correction of the defect. The use of magnetic resonance imaging for the diagnosis of aortopulmonary window has been reported in adults.
The presence of an aortopulmonary window is an indication for surgery. If left untreated, infants die of intractable heart failure or rapidly develop pulmonary vascular obstructive disease. Medical therapy is limited to the preoperative period and for patients with irreversible pulmonary hypertension, which poses the only contraindication to immediate repair of the defect.
PREOPERATIVE MANAGEMENT
Because of the challenges with early diagnosis of aortopulmonary window, it tends to present with symptoms of heart failure. Initial resuscitative efforts for patients presenting in shock are aimed at improving systemic output by limiting excessive pulmonary blood flow and are similar to those used in the patient with single-ventricle anatomy and unobstructed pulmonary blood flow or patients with truncus arteriosus. For the patient with a large aortopulmonary window, this often requires intubation and mechanical ventilation as well as sedation and sometimes neuromuscular blockade to achieve a balanced circulation. The use of hypercapnea and minimizing the fraction
of inspired oxygen (FiO2) will increase the pulmonary vascular resistance, decrease left-to-right shunting, and improve systemic oxygen delivery. Inotropic support may be required. Prostaglandin infusion is necessary to maintain ductal patency in patients with aortopulmonary window and interrupted aortic arch or coarctation. These measures should be successful in restoring systemic output, and the patient should go to surgery without a metabolic acidosis.
of inspired oxygen (FiO2) will increase the pulmonary vascular resistance, decrease left-to-right shunting, and improve systemic oxygen delivery. Inotropic support may be required. Prostaglandin infusion is necessary to maintain ductal patency in patients with aortopulmonary window and interrupted aortic arch or coarctation. These measures should be successful in restoring systemic output, and the patient should go to surgery without a metabolic acidosis.
 SURGICAL TECHNIQUE
SURGICAL TECHNIQUEA median sternotomy incision is used for aortopulmonary window regardless of associated abnormalities. The anatomy should be carefully assessed (Fig. 81.3). The external extent of the aortopulmonary window and the coronary arteries should be identified. Coronary arteries involved in the defect can be seen arising from the area of the communication and coursing down the proximal aorta before reaching the myocardium. The position of the right pulmonary artery should be noted.
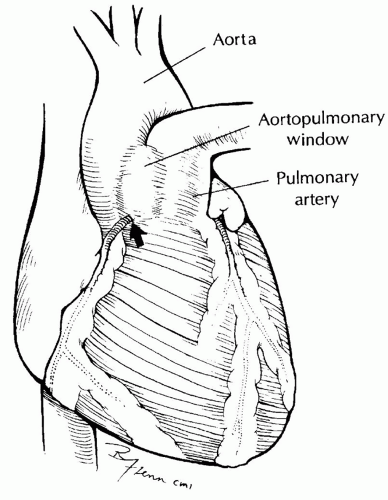 Fig. 81.3. External view of aortopulmonary window. The area of communication between the great vessels can be easily identified. The extent of the defect and the origins of the right pulmonary artery and right coronary artery should be identified. As in this figure, the right coronary artery (arrow) can be sometimes seen originating near the inferior margin of the defect and coursing proximally on the aorta before taking its normal position in the atrioventricular groove. This finding should prompt careful internal inspection for abnormal origin of the coronary artery from the inferior ridge of the defect or the pulmonary artery. |
Simple Aortopulmonary Window
The right and left pulmonary arteries should be loosely encircled with snares so that once cardiopulmonary bypass is established, pulmonary flow can be controlled (Fig. 81.4). General anesthesia can produce a drop in pulmonary vascular resistance, resulting in excessive pulmonary blood flow at the expense of systemic perfusion. It is sometimes helpful to snare one of the branch pulmonary arteries to limit excessive pulmonary blood flow while continuing with preparation for cardiopulmonary bypass. The aorta should be dissected nearly circumferentially distal to the extent of the aortopulmonary window to allow for subsequent placement of the cross-clamp. After the administration of heparin, the aortic cannula is placed in the ascending aorta near the origin of the innominate artery (Fig. 81.4). If there is an associated atrial septal defect or VSD, bicaval cannulation should be undertaken; otherwise, a single venous cannula can be used. Cardiopulmonary bypass is begun and simultaneously the branch pulmonary arteries are snared. A left ventricular vent is placed through the junction of the right superior pulmonary vein and left atrium. A cardioplegia cannula is placed in the ascending aorta. For simple aortopulmonary window, moderate hypothermia to 32°C is adequate. The aorta is cross-clamped distal to the communication. Cardioplegic solution is infused while the pulmonary arteries are snared. The defect can be repaired via an incision in the window itself, through the aorta, or through the pulmonary artery (Fig. 81.5). An approach through the window is preferable because the origin of the coronary arteries can be easily assessed and the patch placed such that an abnormal coronary ostial origin is incorporated into the aorta. In addition, there is less potential for compromise of either of the great vessels or injury of the semilunar valves. The incision is initiated in the anterior-superior portion of the window, and, after the origin of the right coronary artery is identified, the incision is extended proximally, transecting the anterior half of the window. After the origins of the coronary arteries and the right pulmonary artery are identified, an appropriately sized patch of polytetrafluoroethylene (PTFE) or
pericardium is secured to the posterior wall of the defect using continuous suture (Fig. 81.6



pericardium is secured to the posterior wall of the defect using continuous suture (Fig. 81.6
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
