Unstable Angina and Non–ST-Segment Elevation Myocardial Infarction: Introduction
As discussed in detail in Chap. 56, acute coronary syndrome (ACS) is a term used to describe a pattern of clinical symptoms that is consistent with acute myocardial ischemia (Fig. 59–1).1 This chapter discusses two closely related forms of ACS, namely unstable angina (UA) and non–ST-segment elevation myocardial infarction (NSTEMI). NSTEMI (see Fig. 59–1) usually does not progress to Q-wave myocardial infarction (QWMI) but rather to non–Q-wave myocardial infarction (NQMI). Infrequently, NSTEMI evolves to become QWMI by electrocardiography (ECG).
Figure 59–1.

Definitions and pathophysiology of acute coronary syndromes (ACS). Whereas the large majority of patients with ST-segment elevation (large arrows) have complete thrombotic occlusion of an epicardial coronary artery and ultimately develop a Q-wave myocardial infarction (QWMI), a minority (small arrow) develop a non–Q-wave myocardial infarction (NQMI). Patients who present without ST-segment elevation tend to have nonocclusive thrombus and experience either unstable angina or a non–ST-segment elevation myocardial infarction (NSTEMI). Adapted with permission from Davies MJ. Pathophysiology of acute coronary syndromes. Heart 2003;83:361.
The pathophysiology of UA and NSTEMI are very similar, typically involving rupture (or less commonly erosion) of an atherosclerotic plaque with thrombus formation that severely obstructs the coronary artery lumen. Accordingly, patients with either of these syndromes are frequently treated similarly with individual variations in management depending on the classification of patient risk.1 They differ primarily in whether the ischemia is severe enough to lead to a detectable release of a marker of myocardial injury (troponin I, troponin T, or creatine kinase myocardial band [CK-MB]).
Definition and Classification
UA/NSTEMI is also termed non–ST-elevation ACS (NSTE ACS). Angiographic, intravascular ultrasound (IVUS), and angioscopic studies indicate that UA/NSTEMI usually results from the disruption of an atherosclerotic plaque with a subsequent platelet-rich thrombus that obstructs coronary blood flow. The initial diagnosis and management are based on information available at the time of presentation and are updated using new information accumulated over time.1 The initial diagnosis may be challenging because NSTEMI often cannot be differentiated from UA at the time of initial presentation. Moreover, distinguishing UA from the multiple nonischemic causes of chest discomfort may prove difficult and resource intensive.
A patient with symptoms consistent with ACS should have an ECG performed and interpreted within 10 minutes. The most important goal of the early ECG is to identify patients with STEMI who are candidates for immediate reperfusion therapy. Each patient should be given a provisional diagnosis of (1) definite ACS, which should be classified as STEMI, NSTEMI, or UA; (2) possible ACS; (3) a non-ACS cardiac condition (eg, chronic stable angina or heart failure); or (4) a noncardiac diagnosis, which should be as specific as possible. If a provisional diagnosis of ACS is assigned, risk assessment should be performed to determine the probability of major cardiac complications.1 Such risk assessment is not only important among individuals with definite ACS; among patients with possible ACS, risk assessment should be used to determining the contingent probability of an adverse cardiac event if the diagnosis of ACS is confirmed because this information will guide appropriate triage, medical therapy, and timing of subsequent evaluation, including the use of invasive procedures.
Mismatch between myocardial oxygen supply and demand is the cause of myocardial ischemia. Most cases of UA/NSTEMI are attributable to acute reductions in myocardial oxygen supply, which are caused by a platelet-rich thrombus that develops after rupture or erosion of an atherothrombotic plaque (Fig. 59–2). The thrombus may be flow limiting but usually does not completely occlude the epicardial lumen; microembolization of platelet aggregates and components of the disrupted plaque are key components of the acute ischemic syndromes and are likely responsible for the release of biochemical markers of myocardial injury in many patients. A much less common cause (see Fig. 59–2) is dynamic obstruction, which may be a result of intense focal spasm of a segment of an epicardial coronary artery (Prinzmetal or variant angina, as discussed specifically in the section Variant [Prinzmetal] Angina below). UA/NSTEMI may also occur because of severe fixed narrowing of the epicardial coronary artery without spasm or thrombosis, which may occur because of progressive atherosclerosis or restenosis after percutaneous coronary intervention (PCI). In such cases, the presentation may be more gradual than among those with plaque rupture syndromes. These episodes of supply–demand mismatch may occur as a consequence of other clinical events that temporarily increase myocardial oxygen demand beyond what can be supplied by the diseased epicardial coronary arteries. Rarely, UA/NSTEMI may be caused by coronary dissection or vasculitis.
Figure 59–2.
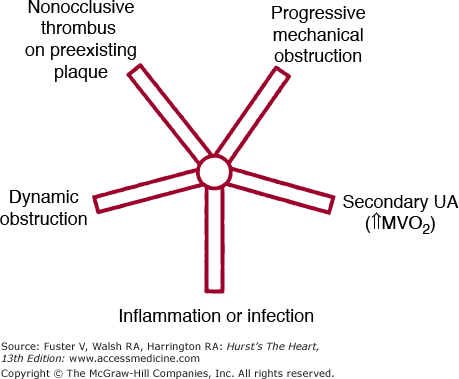
Framework for considering the pathophysiologic components that contribute to unstable angina in a specific patient. Varying contributions are possible from each of the five arms. Some patients have predominantly one cause, but in others, two or more mechanisms contribute significantly. MVO2, myocardial oxygen consumption; UA, unstable angina. Reproduced with permission from Braunwald E. Unstable angina: an etiologic approach to management. Circulation. 1998;98: 2219-2222.
Secondary UA/NSTEMI occurs when the precipitating condition is extrinsic to the coronary arterial bed (see Fig. 59–2). Such patients usually have underlying fixed coronary atherosclerotic narrowing; however, secondary UA/NSTEMI may also be seen in the absence of severe obstructive disease with acute increases in myocardial oxygen demand (eg, fever, tachycardia, hypertensive emergency), reductions in coronary blood flow, (eg, hypotension), or diminution in myocardial oxygen delivery (eg, severe anemia). Among individuals with severe left ventricular (LV) hypertrophy, secondary UA/NSTEMI may occur with a lesser degree of imbalance of oxygen supply and demand.
Initial Presentation
UA may present as rest angina; new-onset severe exertional angina; or angina that is increasing in frequency, duration, or severity. NSTEMI typically presents with rest symptoms that are more prolonged or intense. The different presentations of UA reflect differences in pathophysiology and short-term prognosis, which have implications for therapy. Whereas rest angina more commonly results from an unstable coronary plaque, as described above, new-onset or progressive exertional angina more commonly is caused by fixed atherosclerotic disease without plaque rupture. Other settings include UA/NSTEMI within 6 months after PCI, which is most often caused by restenosis. UA/NSTEMI in a patient with previous coronary bypass surgery often involves advanced atherothrombosis of venous bypass grafts and a lower likelihood of long-term symptomatic relief compared with other patients with UA.
Patients with ACS may present with “atypical” symptoms, which include acute dyspnea, indigestion, unusual locations of pain, agitation, altered mental status, profound weakness, and syncope. Such presentations are more common in women, advanced elderly individuals, and patients with long-standing diabetes mellitus and are associated with higher risk for death and major complications.2 In addition, UA/NSTEMI may be present without evident clinical symptoms, particularly among patients in the perioperative state and those with comorbid medical conditions such as diabetes.
Pathophysiology
| Location of the culprit coronary lesion |
| Stenosis morphology and severity |
| Extent of plaque rupture or erosion |
| Inflammatory substrate |
| Endothelial function |
| Degree of coronary vasoconstriction |
| Microembolization and microvascular obstruction |
| Extent of collaterals |
| Platelet aggregability and reactivity |
| Resistance to antiplatelet agents |
| Leukocyte activation |
| Thrombotic factors and intrinsic clotting activity |
| Level of fibrinolytic activity |
| Blood viscosity |
| Heart rate and blood pressure |
| Catecholamine levels (smoking, cocaine, stress) |
| Blood lipid levels |
| Medical comorbidities (eg, thyroid, renal, pulmonary disease) |
| Compliance with lifestyle and pharmacologic therapies |
Many of the mechanical, cellular, and molecular factors that contribute to plaque disruption have been elucidated in recent years (see Chaps. 52 and 53).3-5 Plaque rupture most commonly occurs at the shoulder region of the plaque, where the plaque joins the adjacent vessel wall; this area of the plaque is commonly infiltrated with inflammatory cells and is subjected to high shear forces.4,5 Plaques prone to rupture (“high-risk” or “vulnerable” plaques) tend to have a thin fibrous cap and a large lipid pool, which influences the biomechanical properties of the plaque and increases the likelihood of rupture (Fig. 59–3). Conversely, fibrous cap thickening appear to decrease the risk of rupture.3,5,6
Figure 59–3.
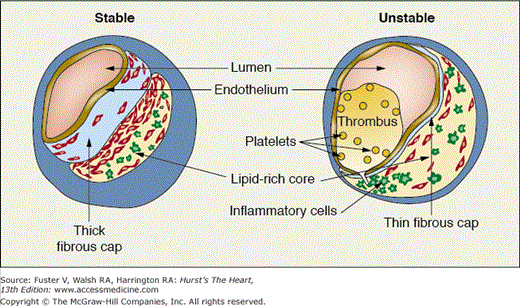
Characteristics of unstable and stable plaque. Although stable plaque may be associated with significant luminal narrowing, it tends to have a thick fibrous cap, a small lipid core, and a paucity of inflammatory cells. In contrast, unstable plaque tends to have a large lipid core and a thin fibrous cap. Inflammatory cells accumulate at the shoulder region and contribute to plaque rupture with subsequent thrombus formation.
Erosion is a less common precipitant of ACS and usually occurs centrally through a thinning cap rather than at the plaque shoulders.3,6 Erosion appears to be more common among women who smoke, but plaque rupture occurs more frequently in hyperlipidemic men.5,6
Inflammation plays a central role in plaque disruption and the subsequent presentation of an ACS (Fig. 59–4). Macrophages and T lymphocytes accumulate in atherothrombotic plaques because of the expression of adhesion molecules on monocytes, endothelial cells, and leukocytes and the release of proinflammatory cytokines and chemokines (eg, monocyte chemoattractant protein-1) that recruit additional inflammatory cells into the region.4
Figure 59–4.

Link between cardiovascular risk factors, endothelial dysfunction, inflammation, and acute coronary syndromes. Endothelial dysfunction may be caused by many proinflammatory atherogenic factors. Endothelial cells thereafter increase expression of adhesion molecules. ACE, angiotensin-converting enzyme; CNP, c-type natriuretic peptide; CRP, C-reactive protein; ICAM, intercellular adhesion molecule; MCP-1, monocyte chemoattractant protein-1; NF, nuclear factor kappa; NO, nitric oxide; PDGF, platelet-derived growth factor; PGI2, prostaglandin; TF, tissue factor; TGF, transforming growth factor; VCAM, vascular cell adhesion molecule.
The matrix metalloproteinases, which include collagenases and gelatinases, are released from macrophages and degrade the collagen that provides strength to the fibrous cap.6 Tissue inhibitors of matrix metalloproteinases are normally expressed by vascular smooth muscle cells. In the vulnerable areas of the fibrous cap, however, macrophages predominate, and smooth muscle cells are sparse, creating an imbalance between matrix-degrading enzymes and their inhibitors.7 It is now believed that in at least in some individuals with UA/NSTEMI, inflammation may be a much more widespread process, reflecting its systemic nature. For example, patients with UA have been demonstrated to have neutrophil activation throughout the coronary tree, including sites remote from the culprit stenosis.8 Moreover, circulating markers of inflammation are elevated in the coronary and systemic circulation of patients with ACS. The clinical manifestations of this phenomenon are now recognized to include the occurrence in some patients with UA/NSTEMI of multiple, simultaneous culprit plaques at the time of presentation.9
The stimuli initiating the acute inflammatory process in ACS have not been clearly delineated. Atherothrombosis, as defined by the “response to injury” hypothesis, is a chronic, low-grade inflammatory condition. Controversy persists as to whether infectious agents play an important role either in atherothrombosis or in the transformation of stable to unstable coronary artery disease (CAD). Chlamydia pneumoniae, cytomegalovirus, and Helicobacter pylori have all been identified within human atherosclerotic lesions, and antibodies to these pathogens are found more often in patients with atherothrombosis than in control subjects. However, these associations do not indicate causality. Moreover, large randomized, controlled trials have failed to show benefit from prolonged treatment with antibiotic regimens that have activity against Chlamydia spp.10,11
Platelet deposition onto the exposed thrombogenic surface of a ruptured plaque is critical to the pathogenesis of UA/NSTEMI. Indeed, a distinguishing feature of thrombosis in the arterial as compared with the venous circulation is the pivotal role of activated platelets. When stimulated by factors such as collagen, epinephrine, adenosine diphosphate (ADP), and thrombin, platelets become activated; undergo a conformational change; and secrete the contents of their α granules, which contain vasoconstrictive substances such as thromboxane A2 and serotonin, as well as procoagulant substances such as fibrinogen and von Willebrand factor. In addition, activation of the platelet causes an increase in surface expression and binding affinity of glycoprotein (GP) IIb/IIIa receptors, which bind fibrinogen and von Willebrand factor to cause platelet aggregation. Activated platelets also release soluble CD40 ligand, which is now recognized as an important immunoregulatory and proinflammatory molecule that may link platelet activation and inflammation.12 Activated platelets and leukocytes interact in the acute phase of UA/NSTEMI to facilitate platelet-thrombus deposition.13
The interplay of activated platelets and leukocytes stimulates the coagulation system, broadly defined as a series of enzymatic events leading to the production of thrombin and the subsequent conversion of fibrinogen to fibrin. Monocytes release tissue factor, a small GP that augments thrombin generation. Tissue factor is also present in the lipid-rich core of atherothrombotic plaques and is likely one of the major determinants of the thrombogenicity of ruptured plaques.3 Tissue factor initiates the extrinsic coagulation cascade, resulting in activation of factor X to factor Xa, which can then convert prothrombin to thrombin. Using phospholipid from the membrane of the activated platelet aggregate, thrombin catalyzes the conversion of fibrinogen to fibrin, forming the platelet-fibrin clot that obstructs coronary blood flow in ACS. Endogenous fibrinolytic activity, mediated by endothelial tissue plasminogen activator (tPA) and antagonized by plasminogen activator inhibitor-1, contribute to the ongoing dynamic “tension between factors promoting platelet-fibrin clot deposition and those encouraging resolution.”
Embolization of platelet-thrombus and plaque material from the site of the ruptured plaque can lead to microvascular obstruction and initiate a cascade of events, including local inflammation and tissue injury, vasoconstriction, and in situ propagation of platelet-leukocyte aggregates. This is an important contributing factor to adverse outcomes in UA/NSTEMI and a target for pharmacotherapy. Troponin elevation appears to identify patients with UA/NSTEMI who are more likely to have microvascular obstruction.14
Culprit lesions in UA/NSTEMI demonstrate an increased response to vasoconstrictor stimuli compared with other coronary artery segments or culprit lesions of patients with stable angina. Vasoconstriction or the lack of appropriate vasodilation probably contribute significantly to the development of ischemic episodes in patients with UA/NSTEMI and is a potential target for therapy.
If a patient with UA/NSTEMI has had a prior coronary angiogram, the culprit lesion has usually progressed markedly since the prior angiogram. The culprit lesion is likely to be asymmetric or eccentric, with a narrow base or neck. Lesions with irregular borders, overhanging edges, or obvious thrombus at angiography are more likely to initiate another cardiac event in the ensuing months.15
Diagnosis
Patients with symptoms suggestive of ACS require a rapid evaluation and expedient decision making, with the intensity of monitoring and therapy determined both by the probability of ACS and the contingent likelihood of a poor outcome if ACS is subsequently confirmed. Because only approximately 15% of patients evaluated in US emergency departments (EDs) with symptoms suggestive of ACS ultimately have an ACS diagnosis confirmed, an organized, efficient, and systematic strategy is needed to avoid exposing patients to risk associated with both the diagnostic procedures as well as the therapies used and unnecessary resource utilization. Formal chest pain or ACS protocols or critical pathways are recommended strategies to provide a systematic approach to the evaluation of possible ACS and to provide greater adherence to guideline recommendations.1
The initial evaluation of a patient with possible UA/NSTEMI is focused on addressing two interrelated questions: (1) How likely are the presenting symptoms and signs to be attributable to an ACS event? and (2) If ACS is confirmed, what is the risk to the patient?1 This early assessment of short-term risk determines the intensity of initial and often subsequent treatment. Determining whether the presentation is attributable to ACS may prove quite challenging in the absence of ECG changes or typical patterns of cardiac biomarker elevation. In a patient with known CAD, typical symptoms are more likely to be caused by myocardial ischemia, especially if the current symptoms are identical to previous episodes when CAD was objectively documented as the cause. However, because the accuracy of descriptive chest pain characteristics is only moderate, a systematic approach to patient evaluation is required.
When UA is suspected in a patient younger than age 50 years, it is particularly important to consider cocaine use. Cocaine can cause coronary vasospasm and thrombosis in addition to its direct effect on altering myocardial oxygen demands through increases in heart rate and blood pressure; it has been implicated as a cause of ACS.16
Patients with ACS often experience discomfort typical of angina, but the episodes are more severe and prolonged; they often occur at rest or with very low levels of exertion. Also, chest discomfort because of UA/NSTEMI is less likely to be relieved by nitroglycerin (NTG) than pain from stable angina. Some patients may have no chest discomfort but present solely with jaw, neck, ear, arm, and epigastric discomfort or with dyspnea in the absence of pain. These should be considered as “anginal equivalents.”
Evaluation of patients with suspected UA/NSTEMI should include the physician’s opinion of the probability of the symptoms being caused by myocardial ischemia, categorizing the presentation into high-, intermediate-, or low-probability categories (Table 59–2).
| High likelihood |
| Known coronary disease (particularly recent PCI) |
| Typical angina reproducing prior documented angina |
| Hemodynamic or ECG changes during pain |
| Dynamic ST-segment elevation or depression of ≥1 mm |
| Marked symmetric T-wave inversion in multiple precordial leads |
| Elevated cardiac enzymes in a rising and falling pattern |
| Intermediate likelihood |
| Absence of high-likelihood features and any of the following: |
| Typical angina in a patient without prior documented angina |
| Atypical anginal symptoms in diabetics or in nondiabetics with two or more other risk factors |
| Male gender |
| Age older than 70 y |
| Extracardiac vascular disease |
| ST depression 0.5-1.0 mm or T-wave inversion of ≥1 mm |
| Low-level troponin elevation that is “flat” and does not rise or fall |
| Low likelihood |
| Absence of high- or intermediate-likelihood features but may have: |
| Chest discomfort reproduced by palpation |
| T waves flat or inverted <1 mm |
| Normal ECG |
Nausea, sweating, or shortness of breath may accompany episodes of chest discomfort. In elderly or diabetic patients, these symptoms may be the only indication that myocardial ischemia is present. Women also present certain challenges with regard to diagnosis. On the one hand, they have a lower probability of underlying significant coronary heart disease than men. On the other hand, women are also more likely than men to have ACS with an “atypical” presentation.2
The diagnostic yield of the 12-lead ECG is enhanced greatly if it can be recorded during an episode of chest discomfort. Although a normal ECG during chest pain does not rule out ACS, it reduces the probability significantly and is a favorable prognostic sign. Transient ST-segment depression of at least 0.5 mm (Fig. 59–5) that appears during chest discomfort and disappears after relief provides objective evidence of transient myocardial ischemia. When it is a constant finding with or without chest pain, it is less specific. A common but nonspecific ECG pattern in patients with UA/NSTEMI are persistent negative T waves over the involved area (Fig. 59–6). Deeply negative T waves across all the precordial (anterior) leads suggest a proximal, severe, left anterior descending coronary artery stenosis as the culprit lesion and is considered a marker of high risk.
Figure 59–5.

Electrocardiogram recorded during an episode of chest pain at rest in a patient with unstable angina. ST-segment depression above 1 mm is present in leads V4 to V6. This abnormality was not present on the baseline tracing. The chest pain and ST-segment depression disappeared promptly after the administration of sublingual nitroglycerin.
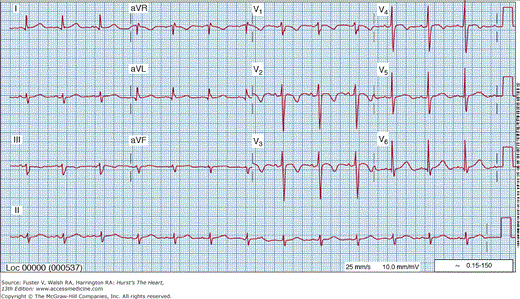
ECG abnormalities may appear or progress in the absence of new symptoms or signs in patients with ACS. Accordingly, it is appropriate and diagnostically useful to obtain serial ECGs during the initial observation period, before discharge, and during recurrent episodes of chest pain.
Biochemical cardiac markers are useful for both the diagnosis of myocardial necrosis and the estimation of prognosis. Cardiac troponins have become the preferred biomarkers for the diagnosis of MI. The absence of cardiac troponins T (cTnT) and I (cTnI) from adult skeletal muscle confers very high cardiac specificity and has allowed the diagnostic threshold for troponin assays to approach zero, resulting in high sensitivity of cTnT and cTnI to detect cardiac injury. As troponin assays become more sensitive, the proportion of individuals with elevation continues to increase; with current assays, more than 30% of individuals with a presentation of UA/NSTEMI have detectable levels of cTnI or cTnT, and with more sensitive assays that will soon be clinically available, the proportion is two- to three-fold higher.17 Many of these will have normal levels of CK-MB. Numerous studies have demonstrated that even minor troponin elevations are independently associated with adverse events in populations with non–ST-elevation ACS.18 It is now clear that patients who have isolated elevation in troponin levels should be classified as having a NSTEMI, provided the clinical syndrome is consistent with ACS. This last caveat is critically important and is discussed further in the below sections. A reflex cardiac evaluation based solely on an elevated cardiac biomarker without considering whether the episode is indeed consistent with an ACS is inappropriate and may be harmful.
Although troponins are highly specific for myocardial injury, they are not specific for the cause of cardiac injury. Multiple acute illnesses besides ACS may cause cardiac injury and lead to troponin elevation (Table 59–3). Particular consideration should be given to the diagnosis of acute pulmonary embolism, which may present with nonspecific chest symptoms and ST- and T-wave changes along with low level elevation in cardiac troponins. Moreover, among individuals with chronic cardiovascular (CV) conditions such as heart failure or chronic CAD, as well as individuals from the general population with diabetes, chronic kidney disease, or asymptomatic LV hypertrophy or LV dysfunction, chronic troponin elevation may be seen, even in the absence of signs and symptoms of ischemia (Fig. 59–7).19,20,22 It is therefore important to interpret troponin values in the context of the clinical presentation and available clinical information.
Figure 59–7.

Troponin T elevation in the general population. Among 3557 participants from the general population enrolled in the Dallas Heart Study, 0.7% had detectable troponin T levels in the absence of signs of cardiac ischemia. Variables associated with troponin elevation included diabetes, left ventricular hypertrophy, evidence of heart failure, and moderate or severe renal insufficiency (left panel). In the absence of one of these criteria, troponin elevation was extremely rare, but the probability of elevated cardiac troponin T (cTnT) increased markedly in proportion to the number of these factors present (right panel). Adapted with permission from Wallace et al.20
| Pulmonary embolus |
| Myocarditis |
| Cardiac contusion |
| Congestive heart failure |
| Chemotherapy (Adriamycin, 5-fluorouracil) |
| Cardioversion or radiofrequency ablation |
| Septic shock |
| Extreme endurance athletics |
| Renal failure |
In most non-ACS conditions associated with cardiac injury, patients with detectable troponin are at increased risk for adverse cardiac outcomes.21,22 However, it should be recognized that in patients with atypical presentations, particularly if they have underlying LV hypertrophy, heart failure, or chronic kidney disease, or diabetes, the elevation in troponin may be caused by a process other than ACS and thus applying an ACS treatment algorithm may be detrimental to the patient because both diagnostic tests and therapies carry potentially adverse consequences such as bleeding. Interpreting troponins in patients with renal failure presents unique problems. Although persistent troponin elevation frequently occurs in patients on hemodialysis even in the absence of signs of ACS, detectable cTnT or cTnI is still associated with very high cardiac event rates.23-25 Knowledge of prior troponin values may help to assess whether a current troponin elevation is new or changing (ie, potentially caused by ACS) or chronic. If ACS is suspected, troponin elevation should be interpreted similarly in patients with and without chronic kidney disease.
As troponin assays become increasingly sensitive, elevations from causes other than ACS are becoming more common and more frustrating for consultant cardiologists. Indeed, recent estimates suggest that as many as 50% of troponin elevations occurring in the hospital setting may be attributable to disease processes other than ACS.26 Newer “high sensitivity” troponin assays have been developed and are expected to be introduced clinically soon; although these assays improve early sensitivity for MI detection,27 they may increase the proportion of elevated levels from causes other than ACS.
At present, few data are available to help clinicians distinguish ACS causes of troponin elevation from other causes. Elevations caused by ACS are usually of a greater magnitude and should demonstrate a dynamic pattern characterized by a transient increase or decrease in values. Demonstration of a transient increase or decrease may improve the specificity for ACS. When a cause other than ACS is suspected, treatment should focus on the underlying disease process. An echocardiogram should be considered, given the frequent association of chronic troponin elevation with cardiac structural abnormalities. Empiric treatment with aspirin (ASA) is recommended if not contraindicated, and in selected individuals, empiric treatment with β-blockers and an evaluation for ischemia may be considered after the acute illness has been stabilized.
The evaluation of patients with chest pain who may have UA or MI is often difficult and uncertain. Hospitalizing all such patients for an extensive workup is unwise and results in unnecessary tests with patient risk and expense. However, missing the diagnosis of MI is the leading cause of malpractice claims for US ED physicians. The chest pain unit was developed as a strategy to prevent missed MI diagnoses while at the same time preventing unnecessary hospitalizations.
Most chest pain units are in or near the ED and are designed to monitor patients with either possible ACS or definite ACS but low-risk features. Criteria usually include chest pain or related symptoms that may indicate myocardial ischemia but with a normal or nondiagnostic ECG and a normal initial set of cardiac enzymes. In most units, cTnT or cTnI is measured at serial intervals (usually every 2-3 hours) for 6 to 9 hours’ duration, sometimes in combination with other serum markers. Some chest pain units measure myoglobin serially over 90 to 180 minutes in a rapid “rule out MI” protocol,28 but this strategy has not been widely adopted. Although some novel biomarkers, including myeloperoxidase and ischemia-modified albumin, have been proposed as tools to risk stratify troponin-negative patients, the value of these additional tests is not clear, particularly as troponin assays become increasingly more sensitive. Recent studies evaluating more sensitive assays for cTnT and cTnI demonstrate improved sensitivity and accuracy for MI detection with these “high-sensitivity” troponin assays, particularly in the first few hours after chest pain presentation.27,29 These assays, which likely will be commercially available soon, may allow shorter protocols for “ruling out” MI using troponin testing alone.
Patients receive an ASA, an intravenous (IV) line, ECG monitoring, and a 12-lead ECG at specific intervals. β-blockers are not recommended if the probability of ACS is considered low and an exercise treadmill test is planned because they may inhibit the heart rate response and decrease the diagnostic yield of the ETT. If cardiac biomarkers remain negative and no ischemia is detected, a symptom-limited stress test may be performed for diagnostic and prognostic purposes or the patient may be discharged and sent home.1 In low-risk patients who present at night or on the weekend, it is reasonable to perform the stress test as an outpatient, provided it can be done within 72 hours.
Intermittent reductions in coronary blood flow distal to the culprit coronary stenosis may occur in patients with UA without either ECG changes or elevation in cardiac biomarkers. Acute rest myocardial perfusion imaging with technetium-labeled sestamibi is a relatively sensitive and specific diagnostic test for ACS. Sestamibi is more useful than thallium for this purpose because imaging can be delayed for up to several hours after injection as a result of minimal redistribution of this imaging agent. ECG-gated images provide an assessment of wall motion in addition to perfusion (see Chap. 21).
The sensitivity and negative predictive value of acute rest perfusion imaging is extremely high if sestamibi is injected during an episode of chest pain, but sensitivity decreases if the injection is done within the ensuing hours. Acute rest imaging is normal in a few (<5%) patients with ACS.
Coronary computed tomography angiography (CTA) is emerging as a potentially useful test among patients with possible ACS (see Chap. 22). This test appears to have high sensitivity, resulting in a high negative predictive value among low- to intermediate-risk patients typically seen in chest pain observation units. In contrast, specificity is a limitation because many lesions that appear obstructive by CTA are nonobstructive with coronary angiography; thus, the positive predictive value is only moderate.30 As such, it represents a potentially useful test when the probability of severe CAD is low, the heart rate can be reduced to 70 beats/min or lower with a β-blocker, and anatomic rather than physiological information is preferred. Radiation exposure is currently an important issue with CTA, although advances in scanner technology and imaging technique are likely to reduce radiation exposure in the future. Current clinical trials are underway comparing a strategy of CTA with functional assessment by various stress test modalities among patients with low- to moderate-risk chest pain syndromes.
Risk Stratification
Initial risk assessment of UA/NSTEMI dictates the appropriate intensity of initial therapy. At the low end of the risk scale, a patient may be discharged home with ASA and an early outpatient stress test. By contrast, high-risk patients may be hospitalized in a coronary care unit, treated with multiple drugs, and undergo coronary angiography urgently as a prelude to revascularization.
The American College of Cardiology (ACC)/American Heart Association 2007 Guideline Update for the Management of Patients with UA and Non–ST-Segment Elevation Myocardial Infarction categorizes patients with UA into low-, intermediate-, and high-risk groups (Table 59–4).1 Features suggesting high risk include ongoing chest pain that lasts longer than 20 minutes, reversible ST-segment changes of at least 0.5 mm, elevated markers of myonecrosis, and signs of significant LV dysfunction. Low-risk patients have worsening angina without rest pain, are age 65 years or younger, have a normal or unchanged ECG without evidence of previous infarction, and have normal cardiac enzymes.
| Feature | High Risk At Least One of the Following Features Must be Present | Intermediate Risk No High Risk Feature, But One or More of the Following Features | Low Risk No High- or Intermediate-Risk Feature But May Have Any of the Following |
|---|---|---|---|
| History | Accelerating tempo of ischemic symptoms in the preceding 48 h | Prior MI, peripheral or cerebrovascular disease, or CABG; prior aspirin use | |
| Character of pain | Prolonged ongoing (>20 min) rest pain | Prolonged (>20 min) rest angina, now resolved, with moderate or high likelihood of CAD Rest angina (<20 min) or relieved with rest or sublingual NTG New-onset or progressive CCS class 3 or 4 angina in the past 2 wk without prolonged (>20 min) rest pain but with moderate or high likelihood of CAD | Increased angina frequency, severity, or duration New-onset angina within 2 wk to 2 mo before presentation |
| Clinical findings | Pulmonary edema, most likely caused by ischemia New or worsening MR murmur S3 or new or worsening rales Hypotension, bradycardia, tachycardia Age older than 75 y | Age older than 70 y | |
| ECG | Angina at rest with transient ST-segment changes >0.05 mV Bundle-branch block, new or presumed new Sustained VT | T-wave inversions >0.2 mV Pathologic Q waves | Normal or unchanged ECG |
| Cardiac markers | Elevated CKMB, cTnT, or cTnI (>0.1 ng/mL) | Slightly elevated CKMB, cTnT, or cTnI (eg, cTnT >0.01 ng/L but <0.1 ng/L) | Normal |
It is increasingly recognized that risk assessment should be both integrative and dynamic. Tools that account for multiple risk predictors provide clear enhancement over evaluation of single variables, as discussed below. Moreover, the risk assessment should be updated during hospitalization. For example, continuing angina with ST-segment changes despite medical therapy is an ominous sign that should prompt urgent coronary arteriography with probable revascularization. Episodes of silent ST-segment depression detected by ambulatory ECG recordings also predict an unfavorable course.
The ECG is too often overlooked as a powerful risk stratification tool in patients with UA/NSTEMI. Even minor ST depression (>0.5 mV) is associated with a markedly increased mortality rate. Among 9461 patients enrolled in the PURSUIT (Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using Integrilin Therapy) study, mortality at 30 days was 5.1% in patients with ST-segment depression versus 2.1% among those without ST-segment depression.31 Also, patients with isolated T-wave inversion have a more favorable prognosis than do those with ST-segment depression.32
Troponin measurements should be used in the risk stratification of patients with UA/NSTEMI to supplement the assessment from clinical and ECG data. Elevated troponin levels strongly predict coronary events over the short term. The combination of troponin elevation and ST-segment depression identifies a group at particularly high risk.33 Coronary angiographic trials have demonstrated that in patients with ACS, troponin elevation is associated with multivessel coronary disease, complex lesion morphology, and visible thrombus, as well as with impairment in microvascular function, which is indicative of distal embolization of plaque material and platelet thrombus to the coronary microcirculation.14 These findings have been confirmed using IVUS34 and explain the consistent association between troponin elevation, even at low levels, and recurrent ischemic events in patients with ACS. The association of troponin elevation with high-risk coronary lesion morphology provides mechanistic insight into studies demonstrating that patients with even minor elevations in cTnT or cTnI derive substantial benefit from an aggressive approach with respect to GPIIb/IIIa inhibitors, low-molecular-weight heparin (LMWH), and an early invasive management strategy.33,35,36
The clinical significance of minor elevations in cardiac troponins has generated controversy, partly because of the many potential causes of troponin elevation besides ACS, as described above, and because of imprecision of troponin assays near the lower end of the assay range. However, recent generations of troponin assays are much more precise at the low end of the measurement range; moreover, studies have consistently demonstrated that patients with minor elevations in troponin values have similar ischemic event rates and derive similar benefit from aggressive therapy as do patients with more markedly elevated troponin values.35 These data argue strongly that any detectable level of troponin with a valid assay should be considered indicative of myocardial injury. When the clinical context is consistent with ACS, the low-level troponin elevations should trigger a more aggressive approach to coronary angiography and antiplatelet therapy.
Plasma levels of B-type natriuretic peptide (BNP) and the N-terminal fragment of its prohormone (NT pro-BNP) may increase in response to cardiac ischemia in proportion to the size and severity of the ischemic insult.37 In patients with UA/NSTEMI, higher levels of BNP or NT pro-BNP measured at presentation or during hospitalization are associated with a markedly increased risk for subsequent death or heart failure, even among patients with normal troponin levels and no evidence of heart failure (Fig. 59–8).38-40 Additionally, it has been demonstrated that serial measurements of BNP provide incremental prognostic information; patients with persistent BNP elevation at 4 or 12 months after ACS have a three- to four-fold increased risk for death at 2 years compared with patients with persistently low BNP levels.41 Despite robust and reproducible prognostic data, the therapeutic implications of isolated BNP or NT pro-BNP elevation in patients with UA/NSTEMI have not been delineated. Thus, currently, selective measurement of BNP and NT pro-BNP is recommended in patients with ACS when further estimation of the risk for death and heart failure is needed, especially more long-term risk.1,42
Figure 59–8.
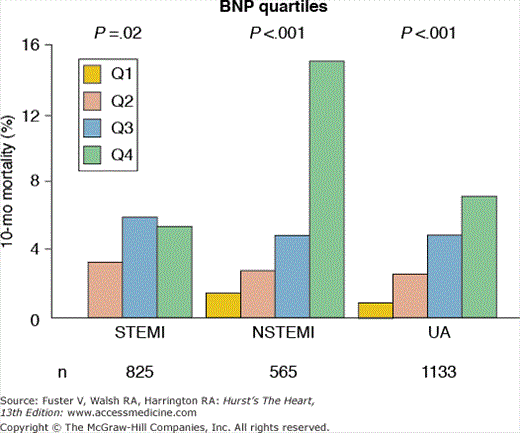
Association between levels of B-type natriuretic peptide (BNP) and mortality across the spectrum of acute coronary syndrome in the OPUS-TIMI 16 study. A robust association between BNP (measured within the first few days after presentation) and 10-month mortality was observed for patients with ST-segment elevation myocardial infarction (STEMI), non–ST-segment elevation myocardial infarction (NSTEMI), and unstable angina (UA). This difference persisted after adjustment for known risk predictors and variables associated with heart failure. OPUS, Orbofiban in Patients with Unstable coronary Syndromes; TIMI, Thrombolysis in Myocardial Infarction. Adapted with permission from de Lemos et al.38
Numerous studies have also evaluated C-reactive protein (CRP) measurement early after ACS, when levels typically increase markedly in response to cardiac injury. For example, in a FRISC (Fragmin During Instability in Coronary Artery Disease) II substudy, CRP levels above 10 mg/L were associated with an increased risk of CV death at all troponin levels.43 However, CRP appears to provide less prognostic information than troponins and BNP/NT pro-BNP. Moreover, the therapeutic implications of early CRP elevation have not been well studied. As such, routine measurement of CRP after an ACS event is not recommended.
Although many biomarkers have been proposed for measurement at various time points after ACS, few have demonstrated the level of robust evidence necessary for consideration of measurement in routine practice, with the exception of troponins and BNP or NT-proBNP.44 In the future, it is possible that multiple biomarker panels incorporating different biomarkers reflecting distinct and nonredundant components of the underlying pathobiology of ACS may emerge as useful tools for risk assessment and therapeutic selection. Proof-in-principle studies have demonstrated that such panels may improve risk stratification compared with standard risk markers and individual biomarkers after ACS.45 However, many logistical challenges remain before these panels can fulfill their potential roles as tools to provide a “personalized” pathophysiology-guided approach to ACS treatment.
Stress testing (Table 59–5) is commonly used for risk assessment in patients with UA/NSTEMI who are managed with an initial conservative (also called an ischemia-guided strategy; see below) strategy. Low-risk and some intermediate-risk patients who stabilize with medical therapy are candidates for stress testing for risk stratification. Stress tests should be symptom limited rather than submaximal, and a stress ECG without adjunctive imaging is appropriate unless baseline ECG abnormalities would preclude adequate interpretation. Those with high-risk findings, such as ST-segment depression at low exercise levels, or large, reversible perfusion defects should undergo coronary arteriography; those with negative or low-risk results can be treated medically. Low-risk patients who complete a stay in a chest pain unit without objective evidence of myocardial ischemia can safely undergo stress testing for diagnosis and prognostic purposes either immediately or as an outpatient within the next 72 hours.1
| High risk (>3% annual mortality rate) |
| 1. Severe resting LV dysfunction (LVEF <0.35) |
| 2. High-risk treadmill (score ≤11) |
| 3. Severe exercise LV dysfunction (exercise LVEF <0.35) |
| 4. Stress-induced large perfusion defect (particularly if anterior) |
| 5. Stress-induced multiple perfusion defects of moderate size |
| 6. Large, fixed perfusion defect with LV dilatation or increased lung uptake |
| 7. Stress-induced moderate perfusion defect with LV dilatation or increased lung uptake (thallium 201) |
| 8. Echocardiographic wall motion abnormality (involving more than two segments) developing at a low dose of dobutamine (≤10 mg/kg/min) or at a low heart rate (≤120 beats/min) |
| 9. Stress echocardiographic evidence of extensive ischemia |
| Intermediate risk (1%-3% annual mortality rate) |
| 1. Mild to moderate resting LV dysfunction (LVEF 0.35-0.49) |
| 2. Intermediate-risk treadmill score (score –11 to +5) |
| 3. Stress-induced moderate perfusion defect without LV dilatation or increased lung intake |
| 4. Limited stress echocardiographic ischemia with a wall motion abnormality only at higher doses or dobutamine involving ≤2 segments |
| Low risk (<1% annual mortality rate) |
| 1. Low-risk treadmill (score ≥+5) |
| 2. Normal or small myocardial perfusion defect at rest or with stress |
| 3. Normal stress echocardiographic wall motion or no change of limited resting wall motion abnormalities during stress |
In patients who are unable to exercise, pharmacologic testing with dipyridamole, adenosine, regadenoson, or dobutamine can be used to provide the stress and sestamibi imaging or echocardiography can be used as a method of assessment (see Chaps. 16, 18, and 21). Stress testing is not needed in patients whose clinical features already put them into a high-risk category; instead, they should proceed directly to coronary arteriography.
Among patients with stable CAD, risk is proportional to the number of vessels with greater than 50% diameter stenosis and the presence and severity of LV dysfunction. However, the relative prognostic impact of the extent of CAD is probably less with ACS because the risk of short-term events is dominated by features of the culprit lesion, such as whether it induces ST-segment depression or troponin release.
Among patients with UA/NSTEMI who undergo arteriography, approximately 25% have one-vessel disease, 25% have two-vessel disease, and 25% have three-vessel disease. Ten percent have significant main stenosis, and the other 15% will have coronary luminal narrowing of less than 50% or normal vessels on arteriography.46 Patients with left main stenosis of at least 50%, or three-vessel disease with LV dysfunction, derive a survival benefit from coronary artery bypass graft (CABG) surgery compared with medical therapy. Importantly, patients with no significant lesions at angiography benefit from a reorientation of their management. Noncardiac causes of chest pain should be considered (including pulmonary embolism), as well as “syndrome X” and variant angina. If the coronaries are completely angiographically normal, antithrombotic and antiplatelet drugs can often be discontinued and the need for antianginal medication reassessed. Patients who are most likely to have no significant lesions at angiography tend to be women with no ECG changes. Nevertheless, the finding of no significant lesions at angiography is usually unanticipated. Importantly, symptomatic patients without significant obstructive CAD seen with angiography may have more severe atherosclerosis detected by IVUS caused by eccentric coronary artery remodeling, which preserves the lumen size. Therefore, in selected patients, more invasive testing at the time of coronary arteriography, including use of IVUS or measurement of coronary flow reserve using adenosine as a vasodilating agent, may be indicated to help better establish the cause of the acute chest pain.
Risk models and scores predict a probability for adverse outcomes based on combinations of clinical, ECG, and laboratory data available at presentation. These tools integrate multiple predictors and thus provide a more comprehensive risk assessment than does focus on a single variable. Whereas the TIMI (Thrombolysis In Myocardial Infarction) and PURSUIT scores were derived from clinical trial databases, the GRACE (Global Registry of Acute Coronary Events) score was derived from a large international registry.31,47,48 Although all three methods discriminate patients at high versus low risk for short- and intermediate-term adverse outcomes, the GRACE model provides better calibration between predicted and observed rates of death.49,50 The TIMI score does not include any measurement of heart failure. The GRACE model was developed in a less-selected patient population and includes renal insufficiency as a variable, which are two potential advantages over the other methods. The major advantage of the TIMI score is that it is a simple integer sum that can be calculated at the bedside without a calculator (Fig. 59–9); PURSUIT and GRACE use weighted averages of multiple risk factors and may require a computer to calculate. None of the models incorporates information from newer biomarkers such as troponin, BNP, or CRP.
Figure 59–9.
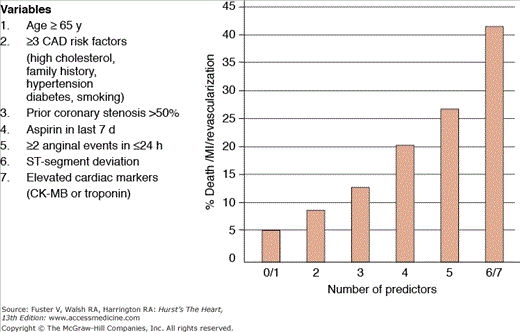
The Thrombolysis in Myocardial Infarction (TIMI) risk score for unstable angina/non–ST-segment elevation myocardial infarction. This simple integer score incorporates variables that can be measured at the bedside and provides both prognostic information as well as a tool to select patients for more or less aggressive treatment algorithms. CAD, coronary artery disease; CK-MB, creatinine kinase myocardial band; MI, myocardial infarction. Adapted with permission from Antman et al.47
In summary, the short-term outcome of UA/NSTEMI can be predicted by a variety of methods (see Table 59–4). The most important of these are the clinical presentation, ECG findings, elevated troponin levels, and continuing episodes of pain despite medical therapy.
Prognosis
Prognosis in patients with UA or UA/NSTEMI depends on the combination of the morbidity or mortality expected from the extent of coronary stenosis and LV dysfunction and the short-term risk associated with the culprit lesion and the unstable state of ACS. Risk is highest early after the onset of symptoms.
Published reports concerning UA/NSTEMI are influenced by patient selection and treatment and can be quite misleading. The inclusion and exclusion criteria for clinical trials introduce bias by often eliminating low- or high-risk patients. Comparisons of incidence and outcome of UA/NSTEMI since the adoption of routine troponin testing are problematic because the proportion of patients classified as having NSTEMI has increased, and the overall mortality risk of NSTEMI may have fallen as a consequence.51
Data from the GRACE study show a 6-month mortality rate of 6.2% in patients with NSTEMI and 3.6% in those with UA. Rehospitalization rates over the 6 month period were approximately 20% and revascularization rates approximately 15%.52



