Background and Historical Perspective
This chapter describes the phenotypic and clinical characteristics of the primary and secondary dilated cardiomyopathies (DCMs), the most common causes of the clinical syndrome of chronic heart failure.1 Heart failure is an enormously important clinical problem, which, if not contained or solved, may ultimately overwhelm health care resources.2 The clinical syndrome of heart failure is a complex process during which the primary pathophysiology is quickly obscured by a variety of superimposed secondary adaptive, maladaptive, and counterregulatory processes (see also Chap. 26). Heart failure is best understood and approached from the vantage point of myocardial failure, most commonly associated with a dilated cardiomyopathy (DCM) phenotype.3 As an indication of their importance, the cardiomyopathies have recently been reclassified by an expert consensus panel under the auspices of the American Heart Association (AHA)4 (see also Chap. 31).
Because of its high prevalence (1%-1.5% of the adult population) and high morbidity, including frequent hospitalizations, the clinical syndrome of heart failure is among the most costly medical problems in the United States.2 Despite improvements in the treatment of heart failure introduced in the past 20 years, including the general availability of cardiac transplantation and better medical treatment, clinical outcome after the onset of symptoms has not changed substantially. The mortality remains high (median survival of 1.7 years for men and 3.2 years for women), the natural history progressive, the cost excessive, and disability and morbidity among the highest of any disease or disease syndrome.1,2,5
Relationship of Myocardial Failure and Dilated Cardiomyopathies to the Clinical Syndrome of Heart Failure
Most cases of heart failure are caused by heart muscle disease (cardiomyopathy). Within the classification of cardiomyopathies (Table 32–1),4,6 the most common cause of the clinical syndrome of heart failure is a secondary (ischemic, valvular, hypertensive, and so on) or a primary (genetic, nongenetic, acquired) DCM, defined as a ventricular chamber exhibiting increased diastolic and systolic volumes and a low (<45%) ejection fraction.7 The natural history of the clinical syndrome of heart failure depends on the course of myocardial failure because (1) the most powerful single predictor of outcome is the degree of left ventricular (LV) dysfunction as assessed by the LV ejection fraction8; (2) treatment that improves intrinsic ventricular function improves the natural history of heart failure3,9; and (3) treatment that ultimately worsens intrinsic function, such as many types of positive inotropic agents, is associated with an adverse effect on outcome.9
| Category | Definition |
|---|---|
| Genetic | |
| I. HCM | ↑↑ Septal and ↑ posterior wall thickness, myofibrillar disarray |
| Mutation in sarcomeric protein, autosomal dominant inheritance | |
| II. ARVC/D | Fibrofatty replacement of RV myocardium |
| III. LV noncompaction | Spongy LV cavity (apex) |
| IV. Glycogen storage diseases | Danon disease, PRKAG2 |
| V. Ion channelopathies | Conduction defects, LQTS, Brugada, SQTS, CPVT, Asian SUNDS |
| Mixed | |
| I. Dilated (DCM) | ↑ EDV ↑ ESV; low EF |
| II. Restrictive (RCM) | ↑ EDV, ↔ ESV; ↑ FP, ↔ EF |
| Acquired | |
| I. Myocarditis | Inflammatory process |
| II. Stress provoked (tako-tsubo) | Reversible LV dysfunction |
| III. Peripartum | Third trimester or 5 months after pregnancy |
| IV. Tachycardia induced | After prolonged periods of SVT or VT |
| V. Infants of insulin-dependent diabetic mothers |
The Classification of Cardiomyopathies
The 1995 World Health Organization/International Society and Federation of Cardiology (WHO/ISFC) classification of cardiomyopathies6 was recently revised to accommodate several rapidly emerging realities, particularly the identification of new disease entities, advances in diagnosis, and knowledge of etiology of previously unknown types of heart muscle disease.4 The classification of cardiomyopathies is discussed in detail in Chaps. 26 and 31.
The new classification of cardiomyopathies is described in Table 32–1. The WHO/ISFC classification of cardiomyopathy was mainly based on the global anatomic description of chamber dimensions in systole and diastole. Thus, the dilated and restrictive categories had definitions based on LV dimensions or volume, which also define function via calculated ejection fraction. The justification for this is that these two groups have distinct natural histories and respond differently to medical treatment. The novel AHA Scientific Statement emphasizes the genetic determinants of cardiomyopathies. Thus, dilated and restrictive cardiomyopathies are defined as mixed cardiomyopathies (predominantly nongenetic); however, hypertrophic cardiomyopathy (HCM), which is caused by mutations in contractile proteins, and other rare forms of cardiomyopathy, including arrhythmogenic right ventricular (RV) cardiomyopathy/dysplasia (ARVC/D) and LV noncompaction (LVNC), which also turned out to be completely genetic in basis, are defined genetic cardiomyopathies. The third category concerns acquired cardiomyopathies, such as peripartum and tachycardia-induced cardiomyopathies. Conversely, genetic cardiomyopathies without unique phenotypes and involvement of a generalized multiorgan disorder, such as the DCM of Becker-Duchenne, are defined as secondary cardiomyopathies. This distinction is arbitrary and may inevitably cause significant overlap between primary and secondary cardiomyopathies.
Finally, the novel classification suggests abandoning the term specific cardiomyopathies and excludes valvular, hypertensive, and ischemic cardiomyopathy from the classification, but many mechanisms responsible for the natural history of myocardial dysfunction are qualitatively similar in primary versus these specific DCM,10 which accurately predicted a qualitatively similar response to treatment targeted at these mechanisms.11,12 In particular, this is the case of ischemic DCM related to previous myocardial infarction (MI) and the subsequent remodeling process, or hypertensive dilated (or restrictive depending on the chamber dimensions) cardiomyopathy, definitions that are still widely used in the clinical practice in the literature.
Molecular Mechanisms in Cardiomyopathies and Myocardial Failure: Disease Phenotype Produced by Alterations in Gene Expression
As shown in Table 32–2, there are three general categories of mechanisms whereby altered gene expression can lead to a phenotypic change in cardiac myocytes:13
A single gene defect, such as lamin A/C gene mutations or α-myosin heavy chain (α-MHC)14-16
Polymorphic variation in modifier genes, such as is present in many components of the renin–angiotensin,17-19 adrenergic,20-23and endothelin systems24
Maladaptive regulated expression of completely normal genes, such as for the mechanisms responsible for progressive myocardial dysfunction and remodeling in secondary DCMs.3,13
| Type of Process | Examples |
|---|---|
| Gene mutation | Cytoskeletal, sarcolemmal, nuclear envelope genes |
| Sarcomeric genes | |
| Signaling pathway genes | |
| Ion channels | |
| Desmosomal genes | |
| Polymorphic variation in modifier genes | ACE, α- and β-adrenergic receptors, endothelin type A receptor |
| Altered expression of a completely normal, wild-type gene (fetal gene program) | Decreased expression: β1-adrenergic receptors, α-MYHC, SERCA2 |
| Increased expression: ANP, β-MYHC, ACE, TNF-α, endothelin, BARK |
The ability to genetically manipulate the cardiovascular system has made it possible to investigate the role of a number of genes in the developing and adult mouse heart (for a review, see Ross25). The discovery that mutations in sarcomeric proteins lead to HCM has made it possible to generate animal models of this disease.26,27 In the case of myosin mutations, a single genetic defect initiates a pathway that ultimately leads to hypertrophy and then, in males, may result in late decompensation and ventricular dilatation.26 Multiple gene mutations have now been associated causally with familial DCMs, as discussed later in this chapter.
A serendipitous genetic model of DCM and heart failure (myf 5 mice) was generated by activation of a skeletal muscle genetic program in the heart.28 These mice have a DCM phenotype characterized by progressive myocardial dysfunction and dilatation. They develop the clinical syndrome of heart failure, and they have an extraordinarily high (>90% at 260 days) heart failure–related mortality. Another serendipitous genetic model of DCM is the muscle LIM protein (MLP) knockout mouse.29 MLP is a positive regulator of muscle differentiation, which is ordinarily expressed at high levels in the heart and may be involved in myofibrillar protein assembly along the actin-based cytoskeleton. MLP knockout mice exhibit typical features of DCM, including decreased systolic and diastolic function and β-adrenergic receptor pathway desensitization.29
These characteristics make this model useful in assessing the mechanisms that lead to the development and progression of myocardial failure. Thus, in transgenic mouse models, both altered expression of contractile proteins and perturbation of myocyte cytoarchitecture can lead to the DCM phenotype.
Several additional transgenic mouse models of cardiomyopathy may be more relevant to the production of a dilated phenotype in humans. Several of them involve overexpression of components of the adrenergic receptor pathway, the heterodimeric G-protein αs subunit (Gαq)30; the α2-,31 β1-,32,33 and β2-adrenergic receptors34; and protein kinase A.35 These β-adrenergic pathway transgenic mouse models exhibit similar histopathology, consisting of myocyte hypertrophy and increased fibrosis, evidence of apoptosis, systolic and diastolic dysfunction, and ultimately development of LV dilatation.30-34,36 Other transgenic mice that ultimately develop a dilated phenotype include those with cardiac-restricted overexpression of activated MEK5,37 CaM kinase IV,38 activated calcineurin,39 and calsequestrin.40 Yet another mouse model of DCM includes mitochondrial transcription factor A gene knockout.41
Several transgenic models of concentric or symmetric LV hypertrophy have been reported, including overexpression of ras,42myc,43 α1-adrenergic receptors,44 the heterodimeric G-protein αq subunit (Gαq),45 and the protein kinase C (PKC).46 The mechanisms for the induction of increased ventricular wall thickness are diverse, inasmuch as the ras, α1-receptor, Gαq, and PKC overexpressors exhibit true cellular hypertrophy with an increase in cell size,42,44-46 whereas the myc animal exhibits cardiac myocyte hyperplasia.43 The HCM phenotypes discussed earlier illustrate the principle that apparently diverse signals can culminate in the same phenotype, presumably by converging on final common pathways.
Multiple gene defects have been identified that can produce DCM in humans, as discussed in more detail in the section on familial forms of DCM. As listed in Table 32–2 and Table 32–3, these include mutations in genes encoding proteins of the cytoskeleton, such as dystrophin47,48; nuclear envelope, such as lamin A/C14,15; sarcomere, such as cardiac β-myosin heavy chain (β-MHC) and α-MHC16,49; ion channels, like SCN5A50,51; desmosome52; and signaling pathways, such transcriptional and Ca2+-cycling regulators.53,54 Likewise, cytoskeletal and sarcomeric gene mutations may cause naturally occurring DCM in animals: in the Syrian hamster, the disease is caused by mutations in the delta-sarcoglycan gene,55 and in turkeys, it is caused by mutations in cardiac troponin T.56
| Phenotype | Estimated Frequency120 (%) | Chromosomal Location | Locus | OMIM | Gene Symbol | Gene |
|---|---|---|---|---|---|---|
| Autosomal dominant familial DCM | 56 | 1q32 | CMD1D | 191045 | TNNT2 | Cardiac troponin T |
| 3p21.1 | 191040 | TNNC1 | Cardiac troponin C | |||
| 2q31 | CMD1G | 188840 | TTN | Titin | ||
| 2q35 | CMD1I | 125660 | DES | Desmin | ||
| 6q12-q16 | CMD1K | 172405 | PLN | Phospholamban | ||
| 9 | CMD1B | 600884 | ||||
| 10q21-q23 | CMD1C | 193065 | VCL | Metavinculin | ||
| 11p11 | 600958 | MYBPC3 | Myosin-binding protein C | |||
| 11p15.1 | CMD1M | 600824 | CSRP3 | Cysteine-glycine–rich protein 3 | ||
| 12q22 | CMD1T | 188380 | LAP2 | Thymopoietin | ||
| 14q12 | CMD1A | 160760 | MYH7 | Cardiac β- MHC | ||
| 14q12 | 160710 | MYH6 | Cardiac α-MHC | |||
| 15q14 | CMD1A | 102540 | ACTC | Cardiac actin | ||
| 15q22.1 | 191010 | TPM1 | α tropomyosin | |||
| 17q12 | CMD1N | 604488 | TCAP | Tinin-cap (teletonin) | ||
| 10q23.2 | 605906 | LDB3 | Cypher/ZASP | |||
| 12p12.1 | 601439 | ABCC9 | Regulatory SUR2A subunit of cardiac KATP channel | |||
| Autosomal recessive familial DCM | 16 | 19q13.42 | 191044 | TNNI3 | Cardiac troponin I | |
| Unknown | 212110 | |||||
| X-linked DCM | 10 | Xp21 | XLCM | 300377 | DMD | Dystrophin |
| Xq24 | 300257 | LAMP2 | Lysosome-associated membrane protein-2 | |||
| Autosomal dominant familial DCM with skeletal muscle disease | 7.7 | 1q11-q23 | LGMD1B | 150330 | LMNA | Lamin A/C |
| 5q33-34 | LGMD2F | 601411 | SGCD | δ-Sarcoglycan | ||
| 4q11 | LGMD2E | 600900 | SGCB | β-Sarcoglycan | ||
| 6q23 | CMD1F | 602067 | ||||
| Autosomal dominant familial DCM with conduction defects | 2.6 | 1q1-q1 | CMD1A | 150330 | LMNA | Lamin A/C |
| 2q14-q22 | CMD1H | 604288 | SCN5A | Na channel, voltage-gated, type V, α-polypetptide | ||
| 3p22.2 | CMD1E | 600163 | ||||
| Rare familial DCM | 7.7 | |||||
| LV noncompaction | Xq28 | 300069 | TAZ | G4.5 (tafazzin) | ||
| 18q12.1-q12.2 | 601239 | DTNA | α-Dystrobrevin | |||
| 10q23.2 | 605906 | LDB3 | Cypher/ZASP | |||
| Autosomal recessive with retinitis pigmentosa and deafness | 6q23-q24 | CMD1J | 605362 | EYA4 | Transcriptional coactivator EYA4 | |
| Autosomal recessive with wooly hair and keratoderma | 6p24 | 125647 | DSP | Desmoplakin | ||
| X-linked congenital DCM | Xq28 | 300069 | TAZ | G4.5 (tafazzin) | ||
| Mitochondrial DCM | mtDNA | 510000 |
Genes exhibit polymorphic variation; for example, normal variants of genes exist in the population that are of slightly different size or sequence.57 Some gene polymorphisms are associated with differences in function of the expressed protein gene product, and some differences in function likely account for the biological variation routinely encountered in population studies of disease susceptibility or clinical response to treatment.
Examples of modifier genes that may have an impact on the natural history of a DCM (see Table 32–2) include the angiotensin-converting enzyme (ACE) DD genotype,17-19 in which individuals are homozygous for the deletion variant, which is associated with increased circulating17 and cardiac tissue58 ACE activity. The DD genotype appears to be a risk factor for early remodeling after MI59 and for the development of end-stage ischemic and idiopathic DCM.18,19 Other potentially important polymorphic variants that may influence the natural history of a cardiomyopathy involve the angiotensin AT1 receptor,60 β2-adrenergic receptors,20 the α2C-adrenergic receptor with or without a β1-receptor polymorphism,21 and the endothelin receptor type A.24
Finally, recent pharmacogenomic studies have shown that polymorphic variations can influence the response to medications. Patients with the DD genotype, who were found to have a worse prognosis, at the same time appeared to respond significantly better to β-blocker therapy than patients with the other genotypes (II and ID).19 Similarly, a polymorphism within a conserved region of the β1-adrenergic receptor (389Arginine) increases the response to isotropic therapy (isoproterenol) and is associated with a reduction of mortality in patients treated with the β-blocker bucindolol.23
The third way for altered gene expression to contribute to the development of a cardiomyopathy is altered, maladaptive expression of a completely normal wild-type gene.13 This occurs most commonly in the context of progression of heart muscle disease and myocardial failure, which is the natural history of virtually all cardiomyopathies after they are established. Examples in this category (see Table 32–2) include downregulation of β1-adrenergic receptors,10 α-MHC,61,62 and the SERCA2 (sarcoplasmic reticulum [SR] Ca2+ adenosine triphosphatase [ATPase])63 genes and upregulation in the atrial natriuretic peptide (ANP),64 β-MHC,61ACE,65,66 tumor necrosis factor alpha (TNF-α),67 endothelin,68 and β-adrenergic receptor kinase (BARK)69 genes. These concepts are discussed further below.
Recent data have shown that in patients who respond to treatment by increasing LV ejection fraction, β-blocker therapy may restore some aspects of altered gene expression, increasing the expression of SR calcium ATPase and of α-MHC and decreasing β-MHC.70
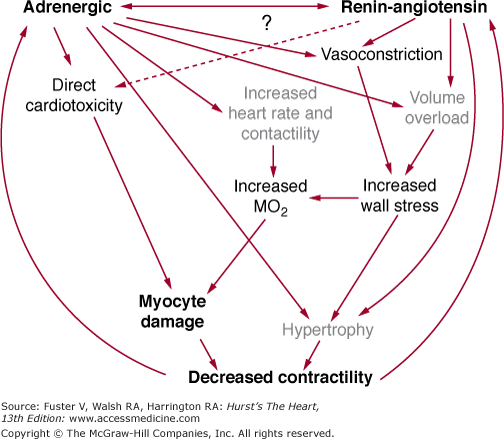
Tissue preparations and myocytes isolated from failing human hearts exhibit evidence of decreased contractile function. Assuming that loading conditions and ischemia are not adversely affecting cardiac myocyte function, in the setting of chronic systolic dysfunction from a DCM, progressive myocardial failure is most likely caused by myocardial cell loss or changes in the gene expression of proteins that regulate or produce muscle contraction. Figures 32–1 and 32–2 summarize these general points and emphasize the central roles of the renin–angiotensin system (RAS) and adrenergic nervous system (ANS) in promoting cell loss, growth and remodeling, and altered gene expression.3
FIGURE 32–1

Relationship of neurohormonal activation and production of cardiac myocyte loss caused by apoptosis and necrosis and altered gene expression. Cell loss and altered gene expression result in more myocardial dysfunction, and a vicious cycle is established. ANS, adrenergic nervous system; RAS, renin–angiotensin system.
Myocardial Dysfunction and Remodeling Caused by Altered Expression of Contractility-Regulating Genes and Changes in Sarcomeric Assembly
Gene expression can be defined broadly as the expression of a fully or normally functioning protein gene product or, more narrowly (and commonly), as the steady-state abundance of a gene’s mRNA transcript. Using either definition, numerous abnormalities of gene expression of normal, wild-type genes have been demonstrated in the failing human heart as discussed earlier, with examples listed in Table 32–2. To characterize the abnormalities that may account for progressive myocardial dysfunction and remodeling, it is useful to subdivide them into two general categories,71 as shown in Table 32–4. The first category encompasses mechanisms that subserve intrinsicfunction, or the mechanisms responsible for contraction and relaxation of the heart in the basal or resting state. Intrinsic function is defined as myocardial contraction and relaxation in the absence of extrinsic influences, such as neurotransmitters or hormones. The second general category is modulatedfunction, which comprises the mechanisms responsible for the remarkable ability of the heart to increase or decrease its performance dramatically (by 2- to 10-fold) and rapidly in response to various physiologic or physical stimuli. Other critical organs such as the brain, kidney, and liver do not exhibit this quality. Modulated function is defined as stimulation or inhibition of myocardial contraction or relaxation by endogenous bioactive compounds, including neurotransmitters, cytokines, autocrine or paracrine substances, and hormones.
| Intrinsic (function in the absence of neural or hormonal influence) | Modulated (function that may be stimulated or inhibited by extrinsic factors including neurotransmitters, cytokines, or hormones) |
|---|---|
| • Contractile proteins | • R-G-phospholipase C pathways |
| • EC coupling mechanisms | • R-G-adenylyl cyclase pathways |
| • R-G-adenylyl cyclase pathways | |
| • Bioenergetics | |
| • Cytoskeleton | |
| • Sarcomere and cell remodeling |
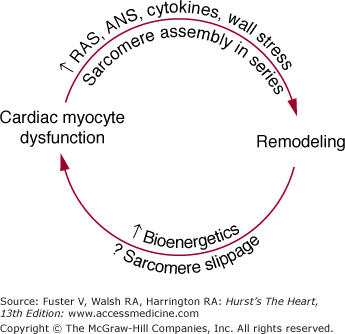
In the failing human heart, changes are present in the expression of genes potentially responsible for both general types of myocardial function depicted in Table 32–4.9,71 Abnormalities of intrinsic function include the factors responsible for an altered length–tension relation,72,73 a blunted force–frequency response,74 or the signals responsible for abnormal cellular and chamber remodeling.3,75 In the case of the abnormal force–frequency and length–tension responses, the evidence favors abnormal contractile function of individual cardiac myocytes. As shown in Table 32–4, these abnormalities likely reside in the contractile proteins or their regulatory elements,61,62 mechanisms involved in excitation–contraction coupling,63 or the cytoskeleton.29,76 However, within these possibilities for altered intrinsic function, there is currently not a consensus as to which specific abnormalities are present in DCM, the most common form of heart failure studied in humans. For cellular remodeling in both human ventricles77 and animal models,78,79 the assembly of sarcomeres in series leads to a myocyte that is markedly increased in length but not in diameter, which contributes to remodeling at the chamber level. Such remodeling places the chamber and the myocyte at an energetic disadvantage because of the attendant increase in wall stress,80 which is one major determinant of myocardial oxygen consumption. Inadequate myocyte energy production, particularly associated with key subcellular ion flux mechanisms or the myosin ATPase cycle,81 in turn contribute to myocyte contractile dysfunction. Moreover, the hypertrophy process itself leads to a qualitative change in contractile protein gene expression (induction of a fetal gene program), which reduces contractile function.13,61,62 Conversely, cardiac myocyte contractile dysfunction likely plays a role in the remodeling process inasmuch as medical treatment, which improves intrinsic myocardial function can reverse remodeling.3,70 Thus, contractile dysfunction and remodeling at the cellular level are intimately related to the progressive contractile dysfunction and chamber enlargement that define the natural history of myocardial failure. These concepts are summarized in Fig. 32–3.
In contrast to abnormalities of intrinsic function, a consensus has been reached on several specific abnormalities in the stimulation component of modulated function. Most changes concern β-adrenergic signal transduction.10,12,71 The ability of β-adrenergic stimulation to increase heart rate and contractility is markedly attenuated in the failing heart caused by multiple changes at the level of receptors, G proteins, and adenylyl cyclase. This produces a major abnormality in the stimulation component of modulated function. In addition, the inhibition component of modulated function is also abnormal in the failing heart because of a reduction in parasympathetic drive.82
There is obviously overlap between the two major subdivisions of myocardial function. Even in the absence of adrenergic stimulation, β-adrenergic receptors have intrinsic activity.83-85 That is, a small percentage of receptors are in an activated state without agonist occupancy and, as such, can support intrinsic myocardial function.84–85 Thus, overexpression of human β2-adrenergic receptors can markedly increase intrinsic myocardial function,84 as can enhancement of SR calcium uptake and release by genetic ablation of the phospholamban gene.86 The realization that active state, agonist-unoccupied β-adrenergic receptors can modulate intrinsic myocardial function is the reason why the R-G-adenylyl cyclase mechanism appears in both categories in Table 32–4.
The second general mechanism by which myocardial function may be adversely affected is by loss of cardiac myocytes, which may also play a role in the progression of ventricular dysfunction in DCMs. Cardiac myocyte loss can occur via toxic mechanisms producing necrosis, or by programmed cell death, producing apoptosis. Apoptosis, which is likely caused by a combination of growth signaling and cell cycle dysregulation, has been described in end-stage DCM87 as well as in the β1-adrenergic receptor,32 in the Gαs-overexpressor transgenic mice,36 and in models of hypertrophy.88 However, data from human studies refer to a very late stage of DCM or ischemic DCM treated with multiple powerful intravenous inotropic medications; therefore, it is unclear whether apoptosis plays a significant role in remodeling or chamber systolic dysfunction (or both) before this point is reached in the natural history of the DCMs.
As depicted in Figs. 32–1 and 32–2, there is now a large body of information supporting the idea that activation of the ANS and RAS compensatory mechanisms contributes to, or is responsible for, the progressive nature of both myocardial failure and the natural history of the heart failure clinical syndrome.9 This evidence includes the observations that activation of both systems is associated with progression of myocardial dysfunction and the heart failure syndrome, and clinical trial data that consistently demonstrate that inhibition of these systems can prevent deterioration in or improve myocardial function as well as reduce mortality.9,12 Although we now know that chronic activation of the ANS and RAS contributes to the progressive nature of myocardial dysfunction in human heart failure, we know virtually nothing about how these systems adversely affect the biology of the cardiac myocyte. What we do know is that mechanisms within both general categories outlined in Table 32–3 must be involved in the adverse myocardial effects mediated by the ANS and RAS. This is so because modulated function may be improved by treatment with ACE inhibitors or β-blocking agents. Progressive myocardial dysfunction and remodeling are attenuated by both β-blocking agents and ACE inhibitors, in cardiomyopathies intrinsic myocardial function is improved and remodeling is reversed by chronic treatment with β-blocking agents.9,70 Additionally, mortality in chronic heart failure is directly related to activation of the ANS89,90 and RAS91 and may be related to the activation of other neurohormonal or autocrine/paracrine systems as well.
Regardless of the type or cause of DCM, an initial myocardial insult resulting in this phenotype exhibits common pathophysiologic features that are summarized in Fig. 32–1. That is, a myocardial insult that produces systolic dysfunction is followed by the initiation of processes designed to temporarily stabilize pump function. The possible mechanisms available for such stabilization are limited. As shown in Fig. 32–2, in chronological order of their action, they are an increase in heart rate and contractility mediated by an increase in cardiac β-adrenergic signaling (produced within seconds of the onset of pump dysfunction), volume expansion to use the Frank-Starling mechanism to increase stroke volume (evident within hours of the onset of pump dysfunction), and cardiac myocyte hypertrophy to increase the number of contractile elements (evident within days or weeks of the onset of pump dysfunction). As shown in Fig. 32–2, these compensatory adjustments are largely accomplished by activation of the RAS and adrenergic nervous ANS systems. However, despite the short-term (days to months) stability achieved via these mechanisms, they ultimately prove harmful.9 The best evidence that chronic, continued activation of the RAS and ANS contributes to progressive myocardial dysfunction and remodeling comes from clinical trials in which both inhibitors of the RAS (ACE inhibitors) and ANS (β-adrenergic receptor–blocking agents) prevent these two phenomena, and β-blocking agents actually may reverse remodeling and progressive systolic dysfunction.3,9
Much current work is focused on the precise pathophysiologic mechanisms by which activation of the RAS and ANS produces remodeling and adverse effects on myocardial function. Some possibilities are given in Fig. 32–1; they include an exacerbation of ischemia or energy depletion, leading to cell loss via necrosis, cell loss by programmed cell death, direct promotion of hypertrophy and remodeling through stimulation of cell growth, and alterations in cardiac myocyte gene expression.3 A key feature of the schema shown in Fig. 32–1 is the process of remodeling, which is discussed in more detail in Chap. 26. Virtually all DCMs undergo this process, which is characterized by progressive dilatation, progressive myocardial systolic dysfunction in viable segments, and a change in chamber shape whereby the ventricle becomes less elliptical and more round.3,9,75 As shown in Fig. 32–3, this places the ventricle at an energetic disadvantage, which likely contributes to further myocardial dysfunction, which then contributes to progressive remodeling. The latter observation is based on data with β-adrenergic blocking agents, which produce an improvement in systolic dysfunction that can be detected prior to a reversal in remodeling.9 As emphasized by Fig. 32–3, each myocardial degenerative process likely begets the other, leading to an inexorably progressive deterioration in myocardial performance and clinical condition.
Scope of Dilated Cardiomyopathies
The number of cardiac or systemic processes that can produce a DCM or are associated with it is plentiful and remarkably varied, as shown in Table 32–5. The dilated phenotype is by far the most common form of cardiomyopathy, comprising more than 90% of subjects referred to specialized centers.92 In the United States, the most common DCM is ischemic DCM,1 or the cardiomyopathy that occurs after MI. Other common secondary DCMs are hypertensive and valvular DCMs, both produced partly by chronically increased wall stress. The primary cardiomyopathy, DCM, is another relatively common dilated phenotype,93,94 as discussed in the following section.
| Ischemic insult (ischemic cardiomyopathy) |
| Valvular disease (mitral regurgitation, aortic stenosis) (valvular cardiomyopathy) |
| Chronic hypertension (hypertensive cardiomyopathy) |
| Tachyarrhythmias (supraventricular, ventricular, atrial flutter) |
| Familial (autosomal dominant, autosomal recessive, X-linked, matrilinear) |
| Idiopathic |
| Toxins |
| Ethanol |
| Chemotherapeutic agents (anthracyclines such as doxorubicin and daunorubicin) |
| Cobalt |
| Antiretroviral agents (zidovudine, didanosine, zalcitabine) |
| Phenothiazines |
| Carbon monoxide |
| Lithium |
| Lead |
| Cocaine |
| Mercury |
| Metabolic abnormalities |
| Nutritional deficiencies (thiamine, selenium, carnitine, protein) |
| Endocrinologic disorders (hypothyroidism, acromegaly, thyrotoxicosis, Cushing’s disease, pheochromocytoma, catecholamines, diabetes mellitus) |
| Electrolyte disturbances (hypocalcemia, hypophosphatemia) |
| Infectious |
| Viral (coxsackievirus, cytomegalovirus, HIV, adenovirus, HSV) |
| Rickettsial |
| Bacterial |
| Mycobacterial |
| Spirochetal |
| Fungal |
| Parasitic (toxoplasmosis, trichinosis, Chagas disease) |
| Autoimmune or collagen disorders |
| Systemic lupus erythematosus |
| Juvenile rheumatoid arthritis |
| Polyarteritis nodosa |
| Kawasaki disease |
| Collagen vascular disorders (scleroderma, lupus erythematosus, dermatomyositis) |
| Infiltrative disorders |
| Hemochromatosis |
| Amyloidosis |
| Sarcoidosis |
| Endomyocardial disorders |
| Hypereosinophilic syndrome (Löffler endocarditis) |
| Endomyocardial fibrosis |
| Hypersensitivity myocarditis |
| Peri- or postpartum dysfunction |
| Arrhythmogenic right ventricular dysplasia or cardiomyopathy |
| Infantile histiocytoid |
| Neuromuscular dystrophies |
| Becker or Duchenne muscular dystrophy, X-linked cardioskeletal myopathy |
| Facioscapulohumeral muscular dystrophy |
| Erb limb-girdle dystrophy |
| Myotonic dystrophy |
| Friedreich ataxia |
| Emery-Dreifuss muscular dystrophy |
| Inborn errors of metabolism |
| Mitochondrial cardiomyopathies |
| Keshan cardiomyopathy |



