THE PATHOGENESIS, PREVENTION, AND TREATMENT OF ATHEROSCLEROSIS
PATHOGENESIS
Atherosclerosis remains the major cause of death and premature disability in developed societies. Moreover, current predictions estimate that by the year 2020 cardiovascular diseases, notably atherosclerosis, will become the leading global cause of total disease burden. Although many generalized or systemic risk factors predispose to its development, atherosclerosis affects various regions of the circulation preferentially and has distinct clinical manifestations that depend on the particular circulatory bed affected. Atherosclerosis of the coronary arteries commonly causes myocardial infarction (MI) (Chap. 35) and angina pectoris (Chap. 33). Atherosclerosis of the arteries supplying the central nervous system frequently provokes strokes and transient cerebral ischemia. In the peripheral circulation, atherosclerosis causes intermittent claudication and gangrene and can jeopardize limb viability. Involvement of the splanchnic circulation can cause mesenteric ischemia. Atherosclerosis can affect the kidneys either directly (e.g., renal artery stenosis) or as a common site of atheroembolic disease (Chap. 38).
Even within a particular arterial bed, stenoses due to atherosclerosis tend to occur focally, typically in certain predisposed regions. In the coronary circulation, for example, the proximal left anterior descending coronary artery exhibits a particular predilection for developing atherosclerotic disease. Similarly, atherosclerosis preferentially affects the proximal portions of the renal arteries and, in the extracranial circulation to the brain, the carotid bifurcation. Indeed, atherosclerotic lesions often form at branching points of arteries which are regions of disturbed blood flow. Not all manifestations of atherosclerosis result from stenotic, occlusive disease. Ectasia and the development of aneurysmal disease, for example, frequently occur in the aorta (Chap. 38). In addition to focal, flow-limiting stenoses, nonocclusive intimal atherosclerosis also occurs diffusely in affected arteries, as shown by intravascular ultrasound and postmortem studies.
Atherogenesis in humans typically occurs over a period of many years, usually many decades. Growth of atherosclerotic plaques probably does not occur in a smooth, linear fashion but discontinuously, with periods of relative quiescence punctuated by periods of rapid evolution. After a generally prolonged “silent” period, atherosclerosis may become clinically manifest. The clinical expressions of atherosclerosis may be chronic, as in the development of stable, effort-induced angina pectoris or predictable and reproducible intermittent claudication. Alternatively, a dramatic acute clinical event such as MI, stroke, or sudden cardiac death may first herald the presence of atherosclerosis. Other individuals may never experience clinical manifestations of arterial disease despite the presence of widespread atherosclerosis demonstrated postmortem.
INITIATION OF ATHEROSCLEROSIS
An integrated view of experimental results in animals and studies of human atherosclerosis suggests that the “fatty streak” represents the initial lesion of atherosclerosis. These early lesions most often seem to arise from focal increases in the content of lipoproteins within regions of the intima. This accumulation of lipoprotein particles may not result simply from increased permeability, or “leakiness,” of the overlying endothelium (Fig. 30-1). Rather, the lipoproteins may collect in the intima of arteries because they bind to constituents of the extracellular matrix, increasing the residence time of the lipid-rich particles within the arterial wall. Lipoproteins that accumulate in the extracellular space of the intima of arteries often associate with glycosaminoglycans of the arterial extracellular matrix, an interaction that may slow the egress of these lipid-rich particles from the intima. Lipoprotein particles in the extracellular space of the intima, particularly those retained by binding to matrix macromolecules, may undergo oxidative modifications. Considerable evidence supports a pathogenic role for products of oxidized lipoproteins in atherogenesis. Lipoproteins sequestered from plasma antioxidants in the extracellular space of the intima become particularly susceptible to oxidative modification, giving rise to hydroperoxides, lysophospholipids, oxysterols, and aldehydic breakdown products of fatty acids and phospholipids. Modifications of the apoprotein moieties may include breaks in the peptide backbone as well as derivatization of certain amino acid residues. Local production of hypochlorous acid by myeloperoxidase associated with inflammatory cells within the plaque yields chlorinated species such as chlorotyrosyl moieties. High-density lipoprotein (HDL) particles modified by HOCl-mediated chlorination function poorly as cholesterol acceptors, a finding that links oxidative stress with impaired reverse cholesterol transport, which is one likely mechanism of the antiatherogenic action of HDL (see later). Considerable evidence supports the presence of such oxidation products in atherosclerotic lesions. A particular member of the phospholipase family, lipoprotein-associated phospholipase A2 (LpPL A2), can generate proinflammatory lipids, including lysophosphatidyl choline-bearing oxidized lipid moieties from oxidized phospholipids found in oxidized low-density lipoproteins (LDLs). An inhibitor of this enzyme is in clinical development.
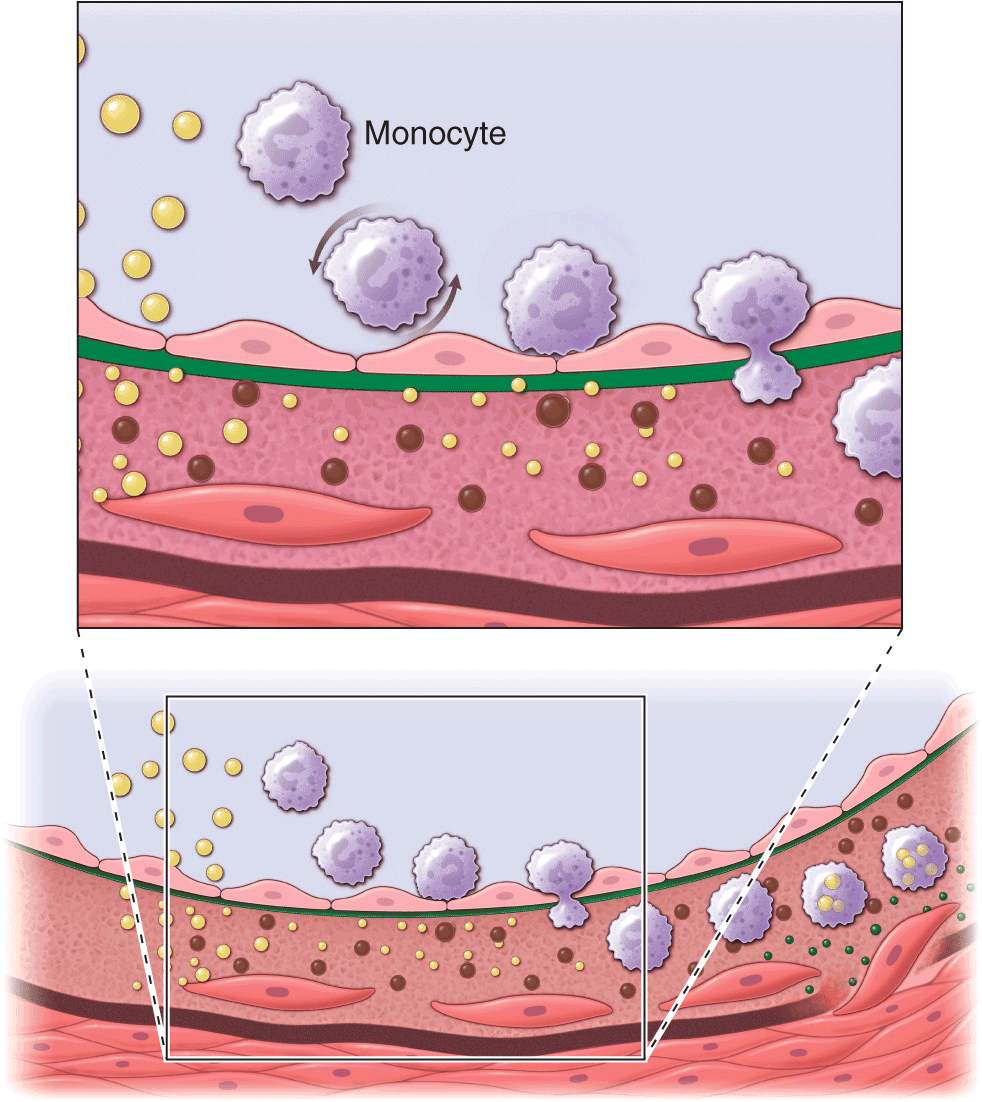
FIGURE 30-1
Cross-sectional view of an artery depicting steps in development of an atheroma, from left to right. The upper panel shows a detail of the boxed area below. The endothelial monolayer overlying the intima contacts blood. Hypercholesterolemia promotes accumulation of LDL particles (light spheres) in the intima. The lipoprotein particles often associate with constituents of the extracellular matrix, notably proteoglycans. Sequestration within the intima separates lipoproteins from some plasma antioxidants and favors oxidative modification. Such modified lipoprotein particles (darker spheres) may trigger a local inflammatory response that signals subsequent steps in lesion formation. The augmented expression of various adhesion molecules for leukocytes recruits monocytes to the site of a nascent arterial lesion.
Once adherent, some white blood cells migrate into the intima. The directed migration of leukocytes probably depends on chemoattractant factors, including modified lipoprotein particles themselves and chemoattractant cytokines (depicted by the smaller spheres), such as the chemokine macrophage chemoattractant protein-1 produced by vascular wall cells in response to modified lipoproteins. Leukocytes in the evolving fatty streak can divide and exhibit augmented expression of receptors for modified lipoproteins (scavenger receptors). These mononuclear phagocytes ingest lipids and become foam cells, represented by a cytoplasm filled with lipid droplets. As the fatty streak evolves into a more complicated atherosclerotic lesion, smooth-muscle cells migrate from the media (bottom of lower panel hairline) through the internal elastic membrane (solid wavy line) and accumulate within the expanding intima, where they lay down extracellular matrix that forms the bulk of the advanced lesion (bottom panel, right side).
Leukocyte recruitment
Accumulation of leukocytes characterizes the formation of early atherosclerotic lesions (Fig. 30-1). Thus, from its very inception, atherogenesis involves elements of inflammation, a process that now provides a unifying theme in the pathogenesis of this disease. The inflammatory cell types typically found in the evolving atheroma include monocyte-derived macrophages and lymphocytes. A number of adhesion molecules or receptors for leukocytes expressed on the surface of the arterial endothelial cell probably participate in the recruitment of leukocytes to the nascent atheroma. Constituents of oxidatively modified low-density lipoprotein can augment the expression of leukocyte adhesion molecules. This example illustrates how the accumulation of lipoproteins in the arterial intima may link mechanistically with leukocyte recruitment, a key event in lesion formation.
Laminar shear forces such as those encountered in most regions of normal arteries also can suppress the expression of leukocyte adhesion molecules. Sites of predilection for atherosclerotic lesions (e.g., branch points) often have disturbed flow. Ordered, pulsatile laminar shear of normal blood flow augments the production of nitric oxide by endothelial cells. This molecule, in addition to its vasodilator properties, can act at the low levels constitutively produced by arterial endothelium as a local anti-inflammatory autacoid, e.g., limiting local adhesion molecule expression. Exposure of endothelial cells to laminar shear stress increases the transcription of Krüppel-like factor 2 (KLF2) and reduces the expression of a thioredoxin-interacting protein (Txnip) that inhibits the activity of the endogenous antioxidant thioredoxin. KLF2 augments the activity of endothelial nitric oxide synthase, and reduced Txnip levels boost the function of thioredoxin. Laminar shear stress also stimulates endothelial cells to produce super-oxide dismutase, an antioxidant enzyme. These examples indicate how hemodynamic forces may influence the cellular events that underlie atherosclerotic lesion initiation and potentially explain the favored localization of atherosclerotic lesions at sites that experience disturbance to laminar shear stress.
Once captured on the surface of the arterial endothelial cell by adhesion receptors, the monocytes and lymphocytes penetrate the endothelial layer and take up residence in the intima. In addition to products of modified lipoproteins, cytokines (protein mediators of inflammation) can regulate the expression of adhesion molecules involved in leukocyte recruitment. For example, interleukin 1 (IL-1) or tumor necrosis factor α (TNF-α) induce or augment the expression of leukocyte adhesion molecules on endothelial cells. Because products of lipoprotein oxidation can induce cytokine release from vascular wall cells, this pathway may provide an additional link between arterial accumulation of lipoproteins and leukocyte recruitment. Chemoattractant cytokines such as monocyte chemoattractant protein 1 appear to direct the migration of leukocytes into the arterial wall.
Foam-cell formation
Once resident within the intima, the mononuclear phagocytes mature into macrophages and become lipid-laden foam cells, a conversion that requires the uptake of lipoprotein particles by receptor-mediated endocytosis. One might suppose that the well-recognized “classic” receptor for LDL mediates this lipid uptake; however, humans or animals lacking effective LDL receptors due to genetic alterations (e.g., familial hypercholesterolemia) have abundant arterial lesions and extraarterial xanthomata rich in macrophage-derived foam cells. In addition, the exogenous cholesterol suppresses expression of the LDL receptor; thus, the level of this cell-surface receptor for LDL decreases under conditions of cholesterol excess. Candidates for alternative receptors that can mediate lipid loading of foam cells include a growing number of macrophage “scavenger” receptors, which preferentially endocytose modified lipoproteins, and other receptors for oxidized LDL or very low-density lipoprotein (VLDL). Monocyte attachment to the endothelium, migration into the intima, and maturation to form lipid-laden macrophages thus represent key steps in the formation of the fatty streak, the precursor of fully formed atherosclerotic plaques.
ATHEROMA EVOLUTION AND COMPLICATIONS
Although the fatty streak commonly precedes the development of a more advanced atherosclerotic plaque, not all fatty streaks progress to form complex atheromata. By ingesting lipids from the extracellular space, the mono-nuclear phagocytes bearing such scavenger receptors may remove lipoproteins from the developing lesion. Some lipid-laden macrophages may leave the artery wall, exporting lipid in the process. Lipid accumulation, and hence the propensity to form an atheroma, ensues if the amount of lipid entering the artery wall exceeds that removed by mononuclear phagocytes or other pathways.
Export by phagocytes may constitute one response to local lipid overload in the evolving lesion. Another mechanism, reverse cholesterol transport mediated by high-density lipoproteins, probably provides an independent pathway for lipid removal from atheroma. This transfer of cholesterol from the cell to the HDL particle involves specialized cell-surface molecules such as the ATP binding cassette (ABC) transporters. ABCA1, the gene mutated in Tangier disease, a condition characterized by very low HDL levels, transfers cholesterol from cells to nascent HDL particles and ABCG1 to mature HDL particles. “Reverse cholesterol transport” mediated by these ABC transporters allows HDL loaded with cholesterol to deliver it to hepatocytes by binding to scavenger receptor B 1 or other receptors. The liver cell can metabolize the sterol to bile acids that can be excreted. This export pathway from macrophage foam cells to peripheral cells such as hepatocytes explains part of the antiatherogenic action of HDLs. (Anti-inflammatory and antioxidant properties also may contribute to the atheroprotective effects of HDLs.) Thus, macrophages may play a vital role in the dynamic economy of lipid accumulation in the arterial wall during atherogenesis.
Some lipid-laden foam cells within the expanding intimal lesion perish. Some foam cells may die as a result of programmed cell death, or apoptosis. This death of mononuclear phagocytes results in the formation of the lipid-rich center, often called the necrotic core, in established atherosclerotic plaques. Macrophages loaded with modified lipoproteins may elaborate cytokines and growth factors that can further signal some of the cellular events in lesion complication. Whereas accumulation of lipid-laden macrophages characterizes the fatty streak, buildup of fibrous tissue formed by extracellular matrix typifies the more advanced atherosclerotic lesion. The smooth-muscle cell synthesizes the bulk of the extracellular matrix of the complex atherosclerotic lesion. A number of growth factors or cytokines elaborated by mononuclear phagocytes can stimulate smooth-muscle cell proliferation and production of extracellular matrix. Cytokines found in the plaque, including IL-1 and TNF-α, can induce local production of growth factors, including forms of platelet-derived growth factor (PDGF), fibroblast growth factors, and others, which may contribute to plaque evolution and complication. Other cytokines, notably interferon γ (IFN-γ) derived from activated T cells within lesions, can limit the synthesis of interstitial forms of collagen by smooth-muscle cells. These examples illustrate how atherogenesis involves a complex mix of mediators that in the balance determines the characteristics of particular lesions.
The arrival of smooth-muscle cells and their elaboration of extracellular matrix probably provide a critical transition, yielding a fibrofatty lesion in place of a simple accumulation of macrophage-derived foam cells. For example, PDGF elaborated by activated platelets, macrophages, and endothelial cells can stimulate the migration of smooth-muscle cells normally resident in the tunica media into the intima. Such growth factors and cytokines produced locally can stimulate the proliferation of resident smooth-muscle cells in the intima as well as those that have migrated from the media. Transforming growth factor β (TGF-β), among other mediators, potently stimulates interstitial collagen production by smooth-muscle cells. These mediators may arise not only from neighboring vascular cells or leukocytes (a “paracrine” pathway), but also, in some instances, may arise from the same cell that responds to the factor (an “autocrine” pathway). Together, these alterations in smooth-muscle cells, signaled by these mediators acting at short distances, can hasten transformation of the fatty streak into a more fibrous smooth-muscle cell and extracellular matrix-rich lesion.
In addition to locally produced mediators, products of blood coagulation and thrombosis likely contribute to atheroma evolution and complication. This involvement justifies the use of the term atherothrombosis to convey the inextricable links between atherosclerosis and thrombosis. Fatty streak formation begins beneath a morphologically intact endothelium. In advanced fatty streaks, however, microscopic breaches in endothelial integrity may occur. Microthrombi rich in platelets can form at such sites of limited endothelial denudation, owing to exposure of the thrombogenic extracellular matrix of the underlying basement membrane. Activated platelets release numerous factors that can promote the fibrotic response, including PDGF and TGF-β. Thrombin not only generates fibrin during coagulation, but also stimulates protease-activated receptors that can signal smooth-muscle migration, proliferation, and extracellular matrix production. Many arterial mural microthrombi resolve without clinical manifestation by a process of local fibrinolysis, resorption, and endothelial repair, yet can lead to lesion progression by stimulating these profibrotic functions of smooth-muscle cells (Fig. 30-2D).
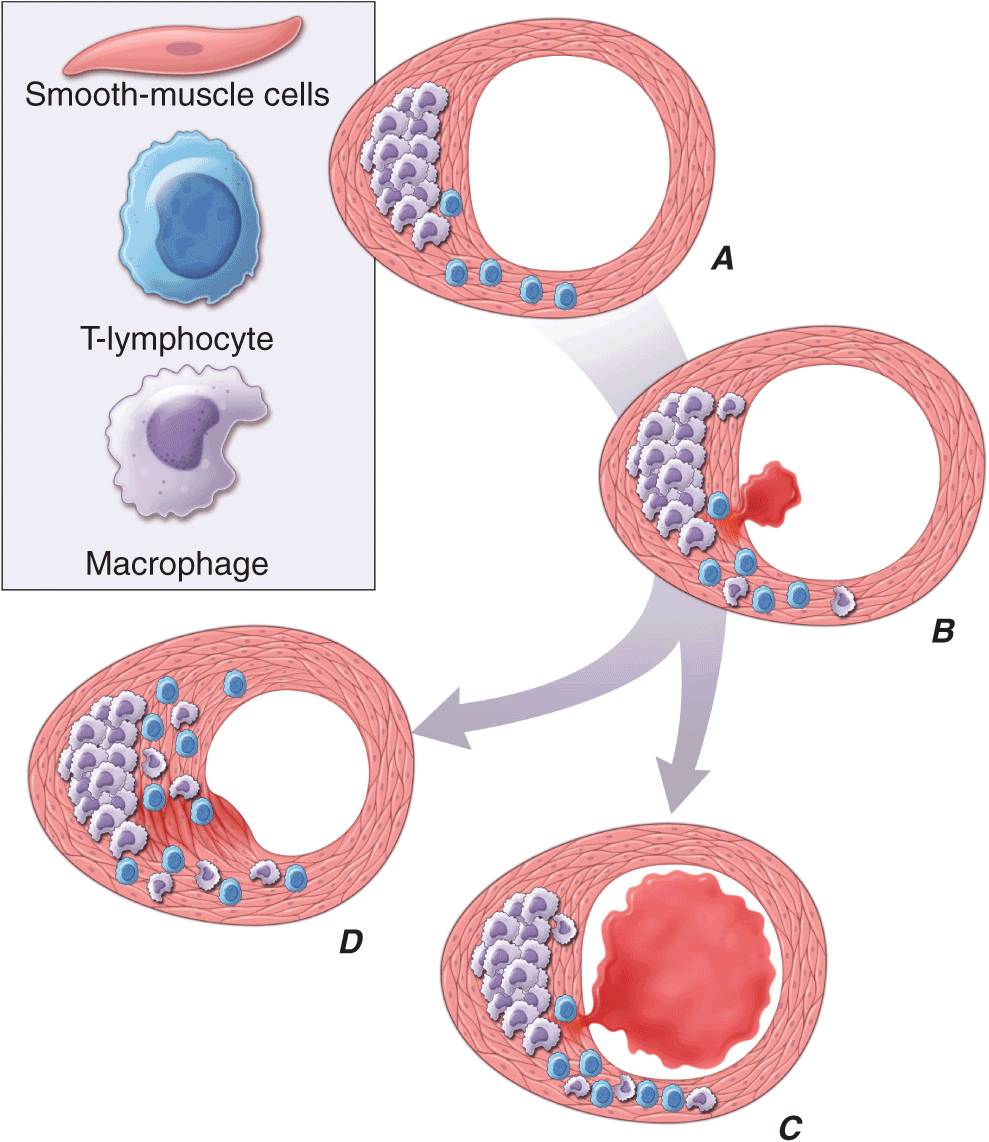
FIGURE 30-2
Plaque rupture, thrombosis, and healing. A. Arterial remodeling during atherogenesis. During the initial part of the life history of an atheroma, growth is often outward, preserving the caliber of the lumen. This phenomenon of “compensatory enlargement” accounts in part for the tendency of coronary arteriography to underestimate the degree of atherosclerosis. B. Rupture of the plaque’s fibrous cap causes thrombosis. Physical disruption of the atherosclerotic plaque commonly causes arterial thrombosis by allowing blood coagulant factors to contact thrombogenic collagen found in the arterial extracellular matrix and tissue factor produced by macrophage-derived foam cells in the lipid core of lesions. In this manner, sites of plaque rupture form the nidus for thrombi. The normal artery wall has several fibrinolytic or antithrombotic mechanisms that tend to resist thrombosis and lyse clots that begin to form in situ. Such antithrombotic or thrombolytic molecules include thrombomodulin, tissue- and urokinase-type plasminogen activators, heparan sulfate proteoglycans, prostacyclin, and nitric oxide. C. When the clot overwhelms the endogenous fibrinolytic mechanisms, it may propagate and lead to arterial occlusion. The consequences of this occlusion depend on the degree of existing collateral vessels. In a patient with chronic multivessel occlusive coronary artery disease (CAD), collateral channels have often formed. In such circumstances, even a total arterial occlusion may not lead to myocardial infarction (MI), or it may produce an unexpectedly modest or a non-ST-segment elevation infarct because of collateral flow. In a patient with less advanced disease and without substantial stenotic lesions to provide a stimulus for collateral vessel formation, sudden plaque rupture and arterial occlusion commonly produces an ST-segment elevation infarction. These are the types of patients who may present with MI or sudden death as a first manifestation of coronary atherosclerosis. In some cases, the thrombus may lyse or organize into a mural thrombus without occluding the vessel. Such instances may be clinically silent. D. The subsequent thrombin-induced fibrosis and healing causes a fibroproliferative response that can lead to a more fibrous lesion that can produce an eccentric plaque that causes a hemodynamically significant stenosis. In this way, a nonocclusive mural thrombus, even if clinically silent or causing unstable angina rather than infarction, can provoke a healing response that can promote lesion fibrosis and luminal encroachment. Such a sequence of events may convert a “vulnerable” atheroma with a thin fibrous cap that is prone to rupture into a more “stable” fibrous plaque with a reinforced cap. Angioplasty of unstable coronary lesions may “stabilize” the lesions by a similar mechanism, producing a wound followed by healing.
Microvessels
As atherosclerotic lesions advance, abundant plexuses of microvessels develop in connection with the artery’s vasa vasorum. Newly developing microvascular networks may contribute to lesion complications in several ways. These blood vessels provide an abundant surface area for leukocyte trafficking and may serve as the portal for entry and exit of white blood cells from the established atheroma. Microvessels in the plaques may also furnish foci for intraplaque hemorrhage. Like the neovessels in the diabetic retina, microvessels in the atheroma may be friable and prone to rupture and can produce focal hemorrhage. Such a vascular leak can provoke thrombosis in situ, yielding local thrombin generation, which in turn can activate smooth-muscle and endothelial cells through ligation of protease-activated receptors. Atherosclerotic plaques often contain fibrin and hemosiderin, an indication that episodes of intraplaque hemorrhage contribute to plaque complications.
 Calcification
Calcification
As they advance, atherosclerotic plaques also accumulate calcium. Proteins usually found in bone also localize in atherosclerotic lesions (e.g., osteocalcin, osteopontin, and bone morphogenetic proteins). Mineralization of the atherosclerotic plaque recapitulates many aspects of bone formation, including the regulatory participation of transcription factors such as Runx2.
Plaque evolution
Although atherosclerosis research has focused much attention on proliferation of smooth-muscle cells, as in the case of macrophages, smooth-muscle cells also can undergo apoptosis in the atherosclerotic plaque. Indeed, complex atheromata often have a mostly fibrous character and lack the cellularity of less advanced lesions. This relative paucity of smooth-muscle cells in advanced atheromata may result from the predominance of cytostatic mediators such as TGF-β and IFN-γ (which can inhibit smooth-muscle cell proliferation), and also from smooth-muscle cell apoptosis. Some of the same proinflammatory cytokines that activate atherogenic functions of vascular wall cells can also sensitize these cells to undergo apoptosis.
Thus, during the evolution of the atherosclerotic plaque, a complex balance between entry and egress of lipoproteins and leukocytes, cell proliferation and cell death, extracellular matrix production, and remodeling, as well as calcification and neovascularization, contribute to lesion formation. Multiple and often competing signals regulate these various cellular events. Many mediators related to atherogenic risk factors, including those derived from lipoproteins, cigarette smoking, and angiotensin II, provoke the production of proinflammatory cytokines and alter the behavior of the intrinsic vascular wall cells and infiltrating leukocytes that underlie the complex pathogenesis of these lesions. Thus, advances in vascular biology have led to increased understanding of the mechanisms that link risk factors to the pathogenesis of atherosclerosis and its complications.
CLINICAL SYNDROMES OF ATHEROSCLEROSIS
Atherosclerotic lesions occur ubiquitously in Western societies. Most atheromata produce no symptoms, and many never cause clinical manifestations. Numerous patients with diffuse atherosclerosis may succumb to unrelated illnesses without ever having experienced a clinically significant manifestation of atherosclerosis. What accounts for this variability in the clinical expression of atherosclerotic disease?
Arterial remodeling during atheroma formation (Fig. 30-2A) represents a frequently overlooked but clinically important feature of lesion evolution. During the initial phases of atheroma development, the plaque usually grows outward, in an abluminal direction. Vessels affected by atherogenesis tend to increase in diameter, a phenomenon known as compensatory enlargement, a type of vascular remodeling. The growing atheroma does not encroach on the arterial lumen until the burden of atherosclerotic plaque exceeds ~40% of the area encompassed by the internal elastic lamina. Thus, during much of its life history, an atheroma will not cause stenosis that can limit tissue perfusion.
Flow-limiting stenoses commonly form later in the history of the plaque. Many such plaques cause stable syndromes such as demand-induced angina pectoris or intermittent claudication in the extremities. In the coronary circulation and other circulations, even total vascular occlusion by an atheroma does not invariably lead to infarction. The hypoxic stimulus of repeated bouts of ischemia characteristically induces formation of collateral vessels in the myocardium, mitigating the consequences of an acute occlusion of an epicardial coronary artery. By contrast, many lesions that cause acute or unstable atherosclerotic syndromes, particularly in the coronary circulation, may arise from atherosclerotic plaques that do not produce a flow-limiting stenosis. Such lesions may produce only minimal luminal irregularities on traditional angiograms and often do not meet the traditional criteria for “significance” by arteriography. Thrombi arising from such nonocclusive stenoses may explain the frequency of MI as an initial manifestation of coronary artery disease (CAD) (in at least one-third of cases) in patients who report no prior history of angina pectoris, a syndrome usually caused by flow-limiting stenoses.
Plaque instability and rupture
Postmortem studies afford considerable insight into the microanatomic substrate underlying the “instability” of plaques that do not cause critical stenoses. A superficial erosion of the endothelium or a frank plaque rupture or fissure usually produces the thrombus that causes episodes of unstable angina pectoris or the occlusive and relatively persistent thrombus that causes acute MI (Fig. 30-2B). In the case of carotid atheromata, a deeper ulceration that provides a nidus for the formation of platelet thrombi may cause transient cerebral ischemic attacks.
Rupture of the plaque’s fibrous cap (Fig. 30-2C) permits contact between coagulation factors in the blood and highly thrombogenic tissue factor expressed by macrophage foam cells in the plaque’s lipid-rich core. If the ensuing thrombus is nonocclusive or transient, the episode of plaque disruption may not cause symptoms or may result in episodic ischemic symptoms such as rest angina. Occlusive thrombi that endure often cause acute MI, particularly in the absence of a well-developed collateral circulation that supplies the affected territory. Repetitive episodes of plaque disruption and healing provide one likely mechanism of transition of the fatty streak to a more complex fibrous lesion (Fig. 30-2D). The healing process in arteries, as in skin wounds, involves the laying down of new extracellular matrix and fibrosis.
Not all atheromata exhibit the same propensity to rupture. Pathologic studies of culprit lesions that have caused acute MI reveal several characteristic features. Plaques that have caused fatal thromboses tend to have thin fibrous caps, relatively large lipid cores, and a high content of macrophages. Morphometric studies of such culprit lesions show that at sites of plaque rupture, macrophages and T lymphocytes predominate and contain relatively few smooth-muscle cells. The cells that concentrate at sites of plaque rupture bear markers of inflammatory activation. In addition, patients with active atherosclerosis and acute coronary syndromes display signs of disseminated inflammation. For example, atherosclerotic plaques and even microvascular endothelial cells at sites remote from the “culprit” lesion of an acute coronary syndrome can exhibit markers of inflammatory activation.
Inflammatory mediators regulate processes that govern the integrity of the plaque’s fibrous cap and, hence, its propensity to rupture. For example, the T cell-derived cytokine IFN-γ, which is found in atherosclerotic plaques, can inhibit growth and collagen synthesis of smooth-muscle cells, as noted earlier. Cytokines derived from activated macrophages and lesional T cells can boost production of proteolytic enzymes that can degrade the extracellular matrix of the plaque’s fibrous cap. Thus, inflammatory mediators can impair the collagen synthesis required for maintenance and repair of the fibrous cap and trigger degradation of extracellular matrix macromolecules, processes that weaken the plaque’s fibrous cap and enhance its susceptibility to rupture (so-called vulnerable plaques). In contrast to plaques with these features of vulnerability, those with a dense extracellular matrix and relatively thick fibrous cap without substantial tissue factor–rich lipid cores seem generally resistant to rupture and unlikely to provoke thrombosis.
Features of the biology of the atheromatous plaque, in addition to its degree of luminal encroachment, influence the clinical manifestations of this disease. This enhanced understanding of plaque biology provides insight into the diverse ways in which atherosclerosis can present clinically and the reasons why the disease may remain silent or stable for prolonged periods, punctuated by acute complications at certain times. Increased understanding of atherogenesis provides new insight into the mechanisms linking it to the risk factors discussed later, indicates the ways in which current therapies may improve outcomes, and suggests new targets for future intervention.
PREVENTION AND TREATMENT
THE CONCEPT OF ATHEROSCLEROTIC RISK FACTORS
The systematic study of risk factors for atherosclerosis emerged from a coalescence of experimental results, as well as from cross-sectional and ultimately longitudinal studies in humans. The prospective, community-based Framingham Heart Study provided rigorous support for the concept that hypercholesterolemia, hypertension, and other factors correlate with cardiovascular risk. Similar observational studies performed worldwide bolstered the concept of “risk factors” for cardiovascular disease.
From a practical viewpoint, the cardiovascular risk factors that have emerged from such studies fall into two categories: those modifiable by lifestyle and/or pharmacotherapy, and those that are immutable, such as age and sex. The weight of evidence supporting various risk factors differs. For example, hypercholesterolemia and hypertension certainly predict coronary risk, but the magnitude of the contributions of other so-called nontraditional risk factors, such as levels of homocysteine, levels of lipoprotein (a) (Lp[a]), and infection, remains controversial. Moreover, some biomarkers that predict cardiovascular risk may not participate in the causal pathway for the disease or its complications. For example, recent genetic studies suggest that C-reactive protein (CRP) does not itself mediate atherogenesis, despite its ability to predict risk. Table 30-1 lists the risk factors recognized by the current National Cholesterol Education Project Adult Treatment Panel III (ATP III). The later sections will consider some of these risk factors and approaches to their modification.
TABLE 30-1
MAJOR RISK FACTORS (EXCLUSIVE OF LDL CHOLESTEROL) THAT MODIFY LDL GOALS

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


