PULMONARY HYPERTENSION
Pulmonary hypertension, an abnormal elevation in pulmonary artery pressure, may be the result of left heart failure, pulmonary parenchymal or vascular disease, thromboembolism, or a combination of these factors. Whether the pulmonary hypertension arises from cardiac, pulmonary, or intrinsic vascular disease, it generally is a feature of advanced disease. Because the causes of pulmonary hypertension are so diverse, it is essential that the etiology underlying the pulmonary hypertension be clearly determined before beginning treatment.
PATHOPHYSIOLOGY
The right ventricle responds to an increase in pulmonary vascular resistance by increasing right ventricular (RV) systolic pressure to preserve cardiac output. In some patients, chronic changes occur in the pulmonary circulation, resulting in progressive remodeling of the vasculature, which can sustain or promote pulmonary hypertension even if the initiating factor is removed.
The ability of the RV to adapt to increased vascular resistance is influenced by several factors, including age and the rapidity of the development of pulmonary hypertension. For example, a large acute pulmonary thromboembolism can result in RV failure and shock, whereas chronic thromboembolic disease of equal severity may result in only mild exercise intolerance. Coexisting hypoxemia can impair the ability of the ventricle to compensate. Studies support the concept that RV failure occurs in pulmonary hypertension when the RV myocardium becomes ischemic as a result of excessive demands and inadequate RV coronary blood flow. The onset of RV failure, often manifest by peripheral edema, is associated with a poor outcome.
DIAGNOSIS
The most common symptom attributable to pulmonary hypertension is exertional dyspnea. Other common symptoms are fatigue, angina pectoris, syncope, near syncope, and peripheral edema.
The physical examination typically reveals increased jugular venous pressure, a reduced carotid pulse, and a palpable RV impulse. Most patients have an increased pulmonic component of the second heart sound, a right-sided fourth heart sound, and tricuspid regurgitation (Chap. 9). Peripheral cyanosis and/or edema tend to occur in later stages of the disease.
Laboratory findings
(Fig. 40-1) The chest x-ray generally shows enlarged central pulmonary arteries. The lung fields may reveal other pathology. The electrocardiogram usually shows right axis deviation and RV hypertrophy. The echocardiogram commonly demonstrates RV and right atrial enlargement, a reduction in left ventricular (LV) cavity size, and a tricuspid regurgitant jet that can be used to estimate RV systolic pressure by Doppler. Pulmonary function tests are helpful in documenting underlying obstructive airways disease, whereas high-resolution chest computed tomography (CT) is preferred to diagnose restrictive lung disease. Hypoxemia and an abnormal diffusing capacity for carbon monoxide occur with pulmonary hypertension of many causes. A perfusion lung scan is almost always abnormal in patients with thromboembolic pulmonary hypertension. However, diffuse defects of a nonsegmental nature often can be seen in long-standing pulmonary hypertension in the absence of thromboemboli. Laboratory tests should include antinuclear antibody and HIV testing. Because of the high frequency of thyroid abnormalities in patients with idiopathic pulmonary hypertension, it is recommended that the thyroid-stimulating hormone level be determined periodically.
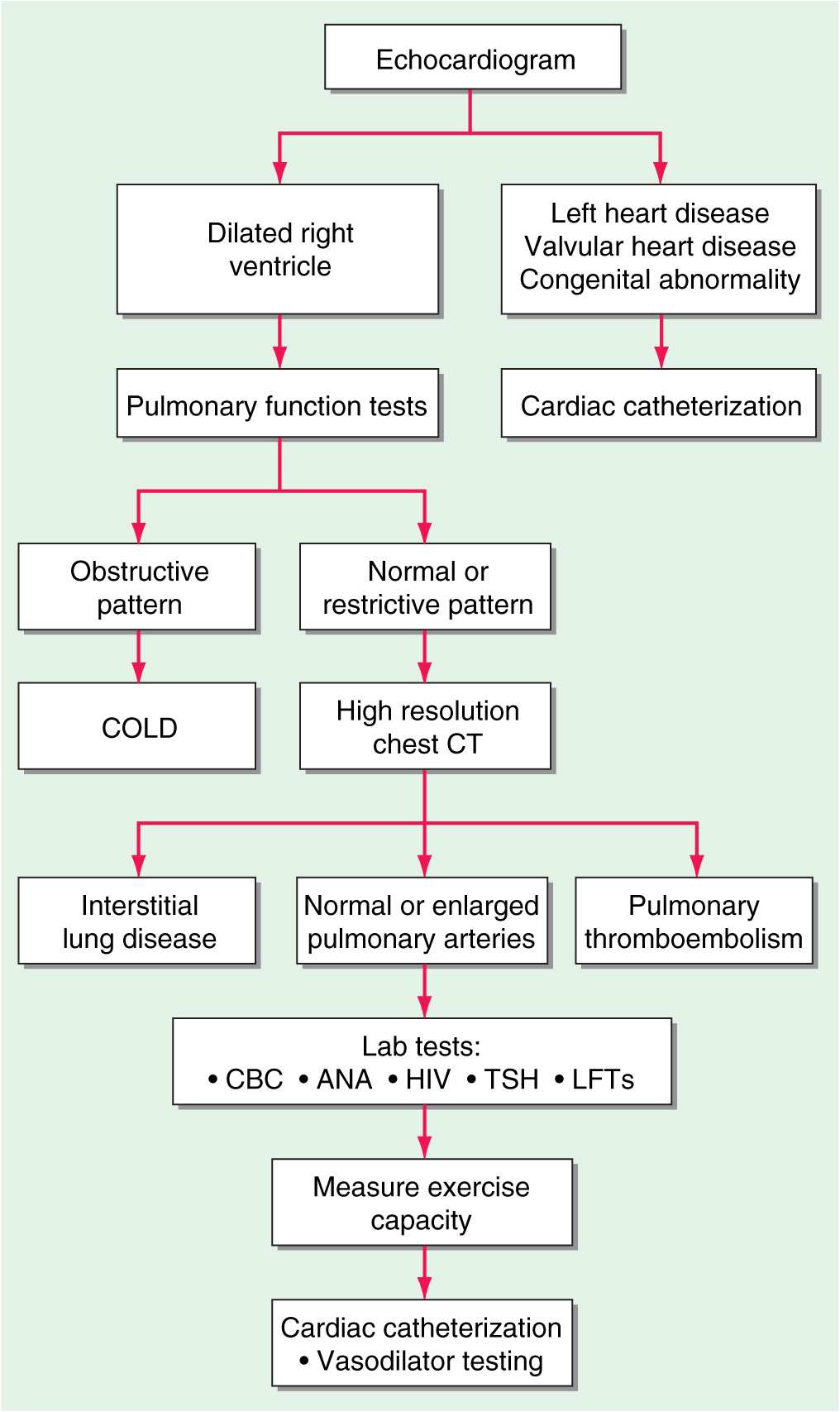
FIGURE 40-1
An algorithm for the workup of a patient with unexplained pulmonary hypertension. All potential etiologies and associated conditions must be investigated in a patient with clinical findings consistent with pulmonary hypertension. COLD, chronic obstructive lung disease; CBC, complete blood count; ANA, antinuclear antibodies; HIV, human immunodeficiency virus; TSH, thyroid-stimulating hormone; LFTs, liver function tests.
Cardiac catheterization
Cardiac catheterization is mandatory for accurate measurement of pulmonary artery pressure, cardiac output, and LV filling pressure as well as documentation of an underlying cardiac shunt. Care should be taken to record pressures only at end expiration. It is recommended that patients with pulmonary arterial hypertension undergo drug testing with a short-acting pulmonary vasodilator to determine the extent of pulmonary vasodilator reactivity. Inhaled nitric oxide, intravenous adenosine, and intravenous epoprostenol have comparable effects in reducing pulmonary artery pressure acutely. Nitric oxide is administered via inhalation in 10–20 parts per million. Adenosine is given in doses of 50 μg/kg per min and increased every 2 min until side effects develop. Epoprostenol is given in doses of 2 ng/kg per min and increased every 30 min until side effects develop. Patients who respond usually can be treated with calcium channel blockers and have a more favorable prognosis.
PULMONARY ARTERIAL HYPERTENSION
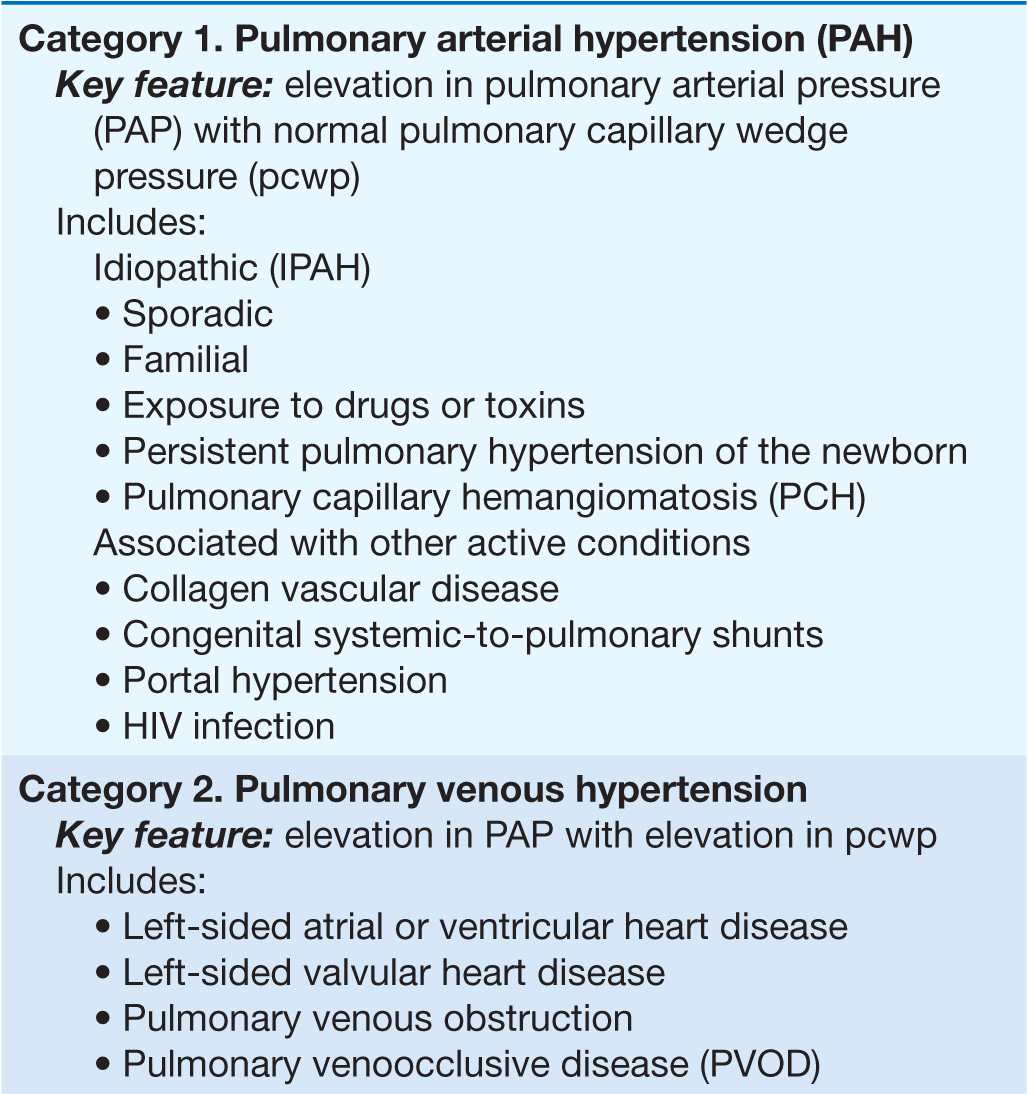
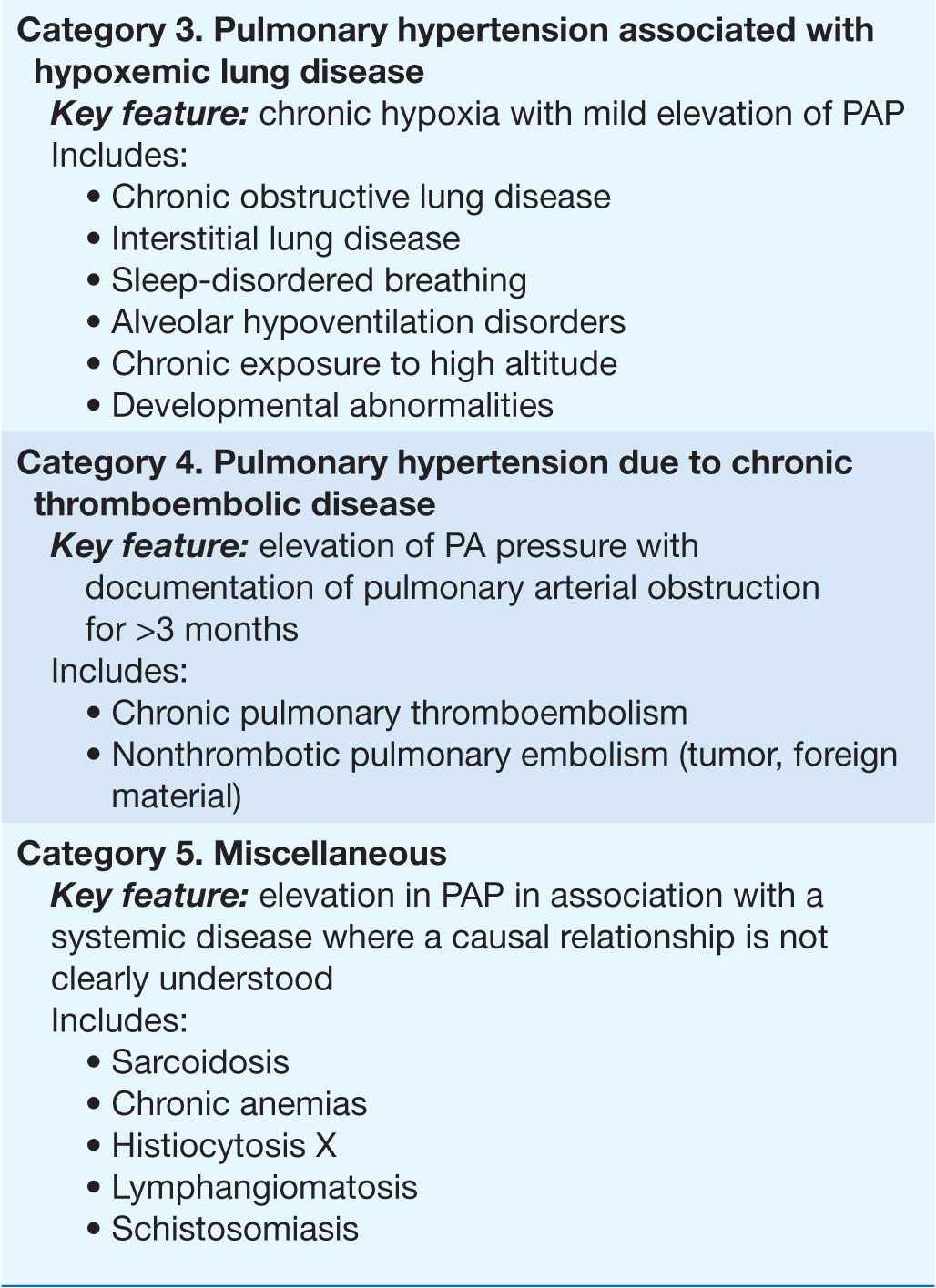
Pulmonary arterial hypertension (PAH) refers to a variety of diseases that include idiopathic PAH, as noted in Table 40-1. Patients with PAH have a common histopathology characterized by medial hypertrophy, eccentric and concentric intimal fibrosis, recanalized thrombi appearing as fibrous webs, and plexiform lesions.
TABLE 40-1
A CLINICAL CLASSIFICATION OF PULMONARY HYPERTENSION
PATHOBIOLOGY
Vasoconstriction, vascular proliferation, thrombosis, and inflammation appear to underlie the development of PAH (Fig. 40-2). Abnormalities in multiple molecular pathways and genes that regulate the pulmonary vascular endothelial and smooth-muscle cells have been identified. These abnormalities include decreased expression of the voltage-regulated potassium channel, mutations in the bone morphogenetic protein-2 receptor, increased tissue factor expression, overactivation of the serotonin transporter, transcription factor activation of hypoxia-inducible factor-1 alpha, and activation of nuclear factor of activated T cells. As a result, there appears to be loss of apoptosis of the smooth-muscle cells that allows their proliferation and the emergence of apoptosis-resistant endothelial cells that can obliterate the vascular lumen. In addition, thrombin deposition in the pulmonary vasculature from a procoagulant state that develops as an independent abnormality or as a result of endothelial dysfunction may amplify the vascular proliferation.
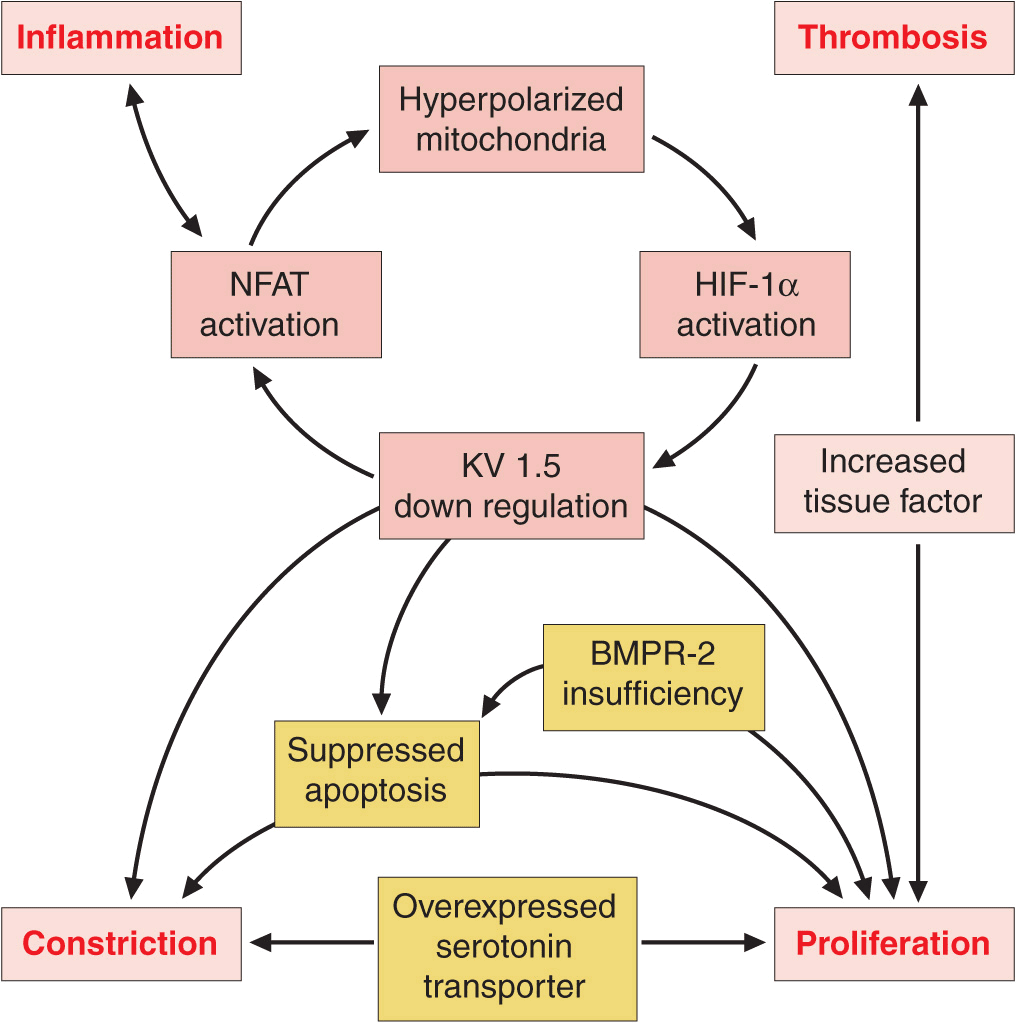
FIGURE 40-2
Multiple biologic pathways that can lead to pulmonary arterial hypertension. Some of the better-characterized ones are illustrated. Because of the redundancy in these pathways and the spectrum of abnormalities that may coexist, it is unlikely that a single agent will produce disease reversal. BMPR-2, bone morphogenetic protein receptor-2; HIF, hypoxia inducible factor; KV 1.5, voltage-regulated potassium channel 1.5; NFAT, nuclear factor of activated T cells.
IDIOPATHIC PULMONARY ARTERIAL HYPERTENSION
Idiopathic pulmonary arterial hypertension (IPAH), formerly referred to as primary pulmonary hypertension, is uncommon, with an estimated incidence of two cases per million. There is a female predominance, with most patients presenting in the fourth and fifth decades, although the age range is from infancy to >60 years.
Familial IPAH accounts for up to 20% of cases of IPAH and is characterized by autosomal dominant inheritance and incomplete penetrance. The clinical and pathologic features of familial and sporadic IPAH are identical. Heterozygous germ-line mutations involving the gene that code the type II bone morphogenetic protein receptor (BMPR II), a member of the transforming growth factor (TGF) β superfamily, appear to account for most cases of familial IPAH. The TGF-β superfami-lies include multifunctional proteins that initiate diverse cellular responses by binding to and activating serine/threonine kinase receptors. The low gene penetrance indicates that other risk factors or abnormalities are necessary to manifest clinical disease. Germ-line mutations in the activin-like kinase gene and endoglin gene, which have been linked to hereditary hemorrhagic telangiectasia, coexist in some patients with familial IPAH.
NATURAL HISTORY
The natural history of IPAH is uncertain, but the disease typically is diagnosed late in its course. Before current therapies, a mean survival of 2–3 years from the time of diagnosis was reported. Functional class remains a strong predictor of survival, with patients who are in New York Heart Association (NYHA) functional class IV having a mean survival of <6 months. The cause of death is usually RV failure, which is manifest by progressive hypoxemia, tachycardia, hypotension, and edema.
TREATMENT Pulmonary Arterial Hypertension
Because the pulmonary artery pressure in PAH increases with exercise, patients should be cautioned against participating in activities that impose physical stress. Diuretic therapy relieves peripheral edema and may be useful in reducing RV volume overload. Pulse oximetry should be monitored, as O2 supplementation helps alleviate dyspnea and RV ischemia in patients whose arterial O2 saturation is reduced. Anticoagulant therapy is advocated for all patients with PAH based on studies demonstrating that warfarin increases survival of patients with PAH. The dose of warfarin generally is titrated to achieve an international normalized ratio (INR) of 2–3 times control.
Several treatments are approved for PAH; they are reviewed below without making a distinction among the different types. However, the efficacy and side effects of these drugs may not be the same in all types of PAH. Other than calcium channel blockers, none of the drugs produce a significant lowering of the pulmonary arterial pressure, and their long-term effects on survival are undefined. The principles for the selection and use of the approved drug treatments are reviewed in Table 40-2.
TABLE 40-2
PRINCIPLES OF DRUG TREATMENT OF PULMONARY ARTERIAL HYPERTENSION

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


