THE BRADYARRHYTHMIAS
David D. Spragg ![]() Gordon F. Tomaselli
Gordon F. Tomaselli
Electrical activation of the heart normally originates in the sinoatrial (SA) node, the predominant pacemaker. Other subsidiary pacemakers in the atrioventricular (AV) node, specialized conducting system, and muscle may initiate electrical activation if the SA node is dysfunctional or suppressed. Typically, subsidiary pacemakers discharge at a slower rate and, in the absence of an appropriate increase in stroke volume, may result in tissue hypoperfusion.
Spontaneous activation and contraction of the heart are a consequence of the specialized pacemaking tissue in these anatomic locales. As described in Chap. 14, action potentials in the heart are regionally heterogeneous. The action potentials in cells isolated from nodal tissue are distinct from those recorded from atrial and ventricular myocytes (Fig. 15-1). The complement of ionic currents present in nodal cells results in a less negative resting membrane potential compared with atrial or ventricular myocytes. Electrical diastole in nodal cells is characterized by slow diastolic depolarization (phase 4), which generates an action potential as the membrane voltage reaches threshold. The action potential upstrokes (phase 0) are slow compared with atrial or ventricular myocytes, being mediated by calcium rather than sodium current. Cells with properties of SA and AV nodal tissue are electrically connected to the remainder of the myocardium by cells with an electrophysiologic phenotype between that of nodal cells and that of atrial or ventricular myocytes. Cells in the SA node exhibit the most rapid phase 4 depolarization and thus are the dominant pacemakers in a normal heart.
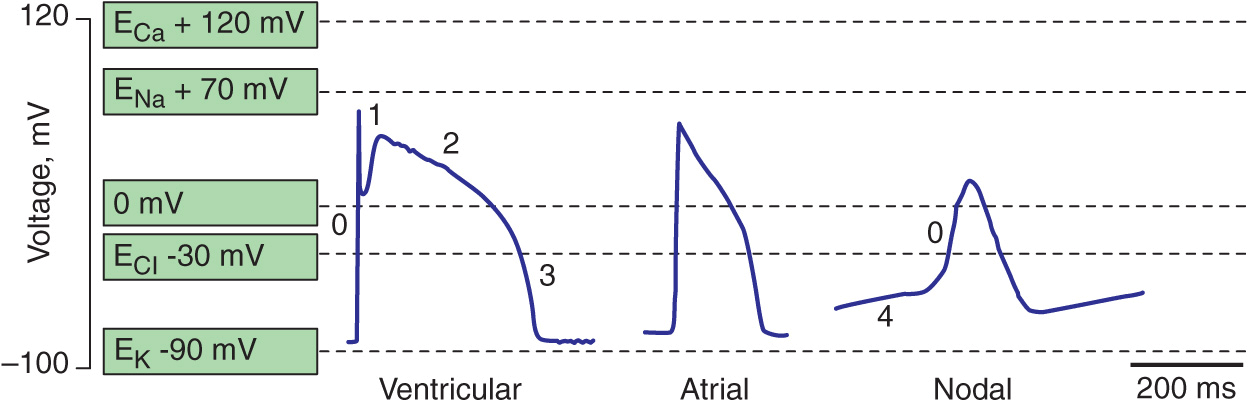
FIGURE 15-1
Action potential profiles recorded in cells isolated from sinoatrial or atrioventricular nodal tissue compared with those of cells from atrial or ventricular myocardium. Nodal cell action potentials exhibit more depolarized resting membrane potentials, slower phase 0 upstrokes, and phase 4 diastolic depolarization.
Bradycardia results from a failure of either impulse initiation or impulse conduction. Failure of impulse initiation may be caused by depressed automaticity resulting from a slowing or failure of phase 4 diastolic depolarization (Fig. 15-2), which may result from disease or exposure to drugs. Prominently, the autonomic nervous system modulates the rate of phase 4 diastolic depolarization and thus the firing rate of both primary (SA node) and subsidiary pacemakers. Failure of conduction of an impulse from nodal tissue to atrial or ventricular myocardium may produce bradycardia as a result of exit block. Conditions that alter the activation and connectivity of cells (e.g., fibrosis) in the heart may result in failure of impulse conduction.
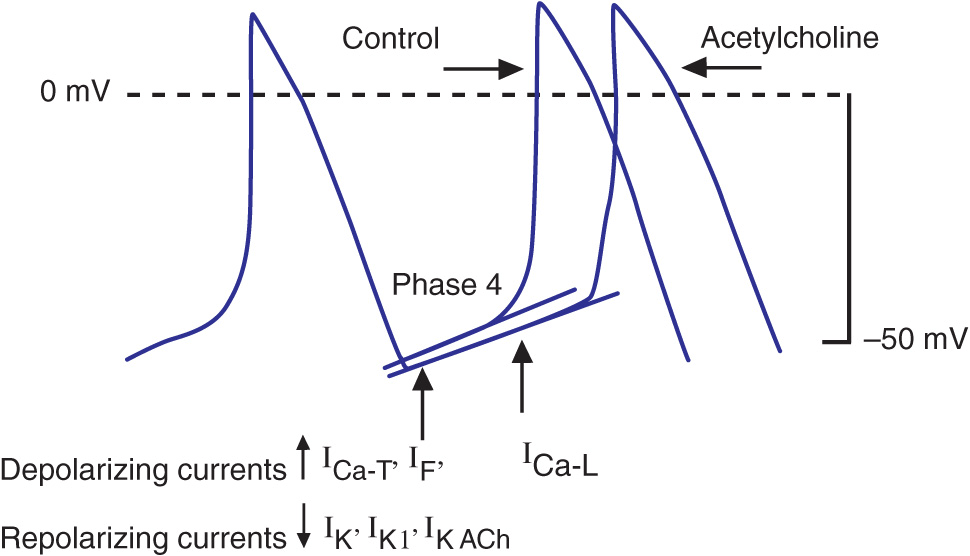
FIGURE 15-2
Schematics of nodal action potentials and the currents that contribute to phase 4 depolarization. Relative increases in depolarizing L- (ICa-L) and T- (ICa-T) type calcium and pacemaker currents (If) along with a reduction in repolarizing inward rectifier (IK1) and delayed rectifier (IK) potassium currents result in depolarization. Activation of ACh-gated (IKACh) potassium current and beta blockade slow the rate of phase 4 and decrease the pacing rate. (Modified from J Jalife et al: Basic Cardiac Electrophysiology for the Clinician, Blackwell Publishing, 1999.)
SA node dysfunction and AV conduction block are the most common causes of pathologic bradycardia. SA node dysfunction may be difficult to distinguish from physiologic sinus bradycardia, particularly in the young. SA node dysfunction increases in frequency between the fifth and sixth decades of life and should be considered in patients with fatigue, exercise intolerance, or syncope and sinus bradycardia. Transient AV block is common in the young and probably is a result of the high vagal tone found in up to 10% of young adults. Acquired and persistent failure of AV conduction is decidedly rare in healthy adult populations, with an estimated incidence of ~200/million population per year.
Permanent pacemaking is the only reliable therapy for symptomatic bradycardia in the absence of extrinsic and reversible etiologies such as increased vagal tone, hypoxia, hypothermia, and drugs (Table 15-1). Approximately 50% of the 150,000 permanent pacemakers implanted in the United States and 20–30% of the 150,000 of those in Europe were implanted for SA node disease.
TABLE 15-1
ETIOLOGIES OF SA NODE DYSFUNCTION

SA NODE DISEASE
Structure and physiology of the SA node
The SA node is composed of a cluster of small fusiform cells in the sulcus terminalis on the epicardial surface of the heart at the right atrial–superior vena caval junction, where they envelop the SA nodal artery. The SA node is structurally heterogeneous, but the central prototypic nodal cells have fewer distinct myofibrils than does the surrounding atrial myocardium, no intercalated disks visible on light microscopy, a poorly developed sarcoplasmic reticulum, and no T-tubules. Cells in the peripheral regions of the SA node are transitional in both structure and function. The SA nodal artery arises from the right coronary artery in 55–60% and the left circumflex artery in 40–45% of persons. The SA node is richly innervated by sympathetic and parasympathetic nerves and ganglia.
Irregular and slow propagation of impulses from the SA node can be explained by the electrophysiology of nodal cells and the structure of the SA node itself. The action potentials of SA nodal cells are characterized by a relatively depolarized membrane potential (Fig. 15-1) of –40 to –60 mV, slow phase 0 upstroke, and relatively rapid phase 4 diastolic depolarization compared with the action potentials recorded in cardiac muscle cells. The relative absence of inward rectifier potassium current (IK1) accounts for the depolarized membrane potential; the slow upstroke of phase 0 results from the absence of available fast sodium current (INa) and is mediated by L-type calcium current (ICa-L); and phase 4 depolarization is a result of the aggregate activity of a number of ionic currents. Prominently, both L- and T-type (ICa-T) calcium currents, the pacemaker current (so-called funny current, or If) formed by the tetramerization of hyperpolarization-activated cyclic nucleotide-gated channels, and the electrogenic sodium-calcium exchanger provide depolarizing current that is antagonized by delayed rectifier (IKr) and acetylcholine-gated (IKACh) potassium currents. ICa-L, ICa-T, and If are modulated by β-adrenergic stimulation and IKACh by vagal stimulation, explaining the exquisite sensitivity of diastolic depolarization to autonomic nervous system activity. The slow conduction within the SA node is explained by the absence of INa and poor electrical coupling of cells in the node, resulting from sizable amounts of interstitial tissue and a low abundance of gap junctions. The poor coupling allows for graded electrophysiologic properties within the node, with the peripheral transitional cells being silenced by electrotonic coupling to atrial myocardium.
Etiology of SA nodal disease
SA nodal dysfunction has been classified as intrinsic or extrinsic. The distinction is important because extrinsic dysfunction is often reversible and generally should be corrected before pacemaker therapy is considered (Table 15-1). The most common causes of extrinsic SA node dysfunction are drugs and autonomic nervous system influences that suppress automaticity and/or compromise conduction. Other extrinsic causes include hypothyroidism, sleep apnea, and conditions likely to occur in critically ill patients such as hypothermia, hypoxia, increased intracranial pressure (Cushing’s response), and endotracheal suctioning via activation of the vagus nerve.
Intrinsic sinus node dysfunction is degenerative and often is characterized pathologically by fibrous replacement of the SA node or its connections to the atrium. Acute and chronic coronary artery disease (CAD) may be associated with SA node dysfunction, although in the setting of acute myocardial infarction (MI; typically inferior), the vabnormalities are transient. Inflammatory processes may alter SA node function, ultimately producing replacement fibrosis. Pericarditis, myocarditis, and rheumatic heart disease have been associated with SA nodal disease with sinus bradycardia, sinus arrest, and exit block. Carditis associated with systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and mixed connective tissue disorders (MCTDs) may also affect SA node structure and function. Senile amyloidosis is an infiltrative disorder in patients typically in the ninth decade of life; deposition of amyloid protein in the atrial myocardium can impair SA node function. Some SA node disease is iatrogenic and results from direct injury to the SA node during cardiothoracic surgery.
Rare heritable forms of sinus node disease have been described, and several have been characterized genetically. Autosomal dominant sinus node dysfunction in conjunction with supraventricular tachycardia (i.e., tachycardia-bradycardia variant of sick-sinus syndrome [SSS]) has been linked to mutations in the pacemaker current (If) subunit gene HCN4 on chromosome 15. An autosomal recessive form of SSS with the prominent feature of atrial inexcitability and absence of P waves on the electrocardiogram (ECG) is caused by mutations in the cardiac sodium channel gene, SCN5A, on chromosome 3. SSS associated with myopia has been described but not genetically characterized. There are several neuromuscular diseases, including Kearns-Sayre syndrome (ophthalmoplegia, pigmentary degeneration of the retina, and cardiomyopathy) and myotonic dystrophy, that have a predilection for the conducting system and SA node.
SSS in both the young and the elderly is associated with an increase in fibrous tissue in the SA node. The onset of SSS may be hastened by coexisting disease, such as CAD, diabetes mellitus, hypertension, and valvular diseases and cardiomyopathies.
Clinical features of SA node disease
SA node dysfunction may be completely asymptomatic and manifest as an ECG anomaly such as sinus bradycardia; sinus arrest and exit block; or alternating supraventricular tachycardia, usually atrial fibrillation, and bradycardia. Symptoms associated with SA node dysfunction, in particular tachycardia-bradycardia syndrome, may be related to both slow and fast heart rates. For example, tachycardia may be associated with palpitations, angina pectoris, and heart failure, and bradycardia may be associated with hypotension, syncope, presyncope, fatigue, and weakness. In the setting of SSS, overdrive suppression of the SA node may result in prolonged pauses and syncope upon termination of the tachycardia. In many cases, symptoms associated with SA node dysfunction result from concomitant cardiovascular disease. A significant minority of patients with SSS develop signs and symptoms of heart failure that may be related to slow or fast heart rates.
One-third to one-half of patients with SA node dysfunction develop supraventricular tachycardia, usually atrial fibrillation or atrial flutter. The incidence of persistent atrial fibrillation in patients with SA node dysfunction increases with advanced age, hypertension, diabetes mellitus, left ventricular dilation, valvular heart disease, and ventricular pacing. Remarkably, some symptomatic patients may experience an improvement in symptoms with the development of atrial fibrillation, presumably from an increase in their average heart rate. Patients with the tachycardia-bradycardia variant of SSS, similar to patients with atrial fibrillation, are at risk for thromboembolism, and those at greatest risk, including patients ≥65 years and patients with a prior history of stroke, valvular heart disease, left ventricular dysfunction, or atrial enlargement, should be treated with anticoagulants. Up to one-quarter of patients with SA node disease will have concurrent AV conduction disease, although only a minority will require specific therapy for high-grade AV block.
The natural history of SA node dysfunction is one of varying intensity of symptoms even in patients who present with syncope. Symptoms related to SA node dysfunction may be significant, but overall mortality usually is not compromised in the absence of other significant comorbid conditions. These features of the natural history need to be taken into account in considering therapy for these patients.
Electrocardiography of SA node disease
The electrocardiographic manifestations of SA node dysfunction include sinus bradycardia, sinus pauses, sinus arrest, sinus exit block, tachycardia (in SSS), and chronotropic incompetence. It is often difficult to distinguish pathologic from physiologic sinus bradycardia. By definition, sinus bradycardia is a rhythm driven by the SA node with a rate of <60 beats/min; sinus bradycardia is very common and typically benign. Resting heart rates <60 beats/min are very common in young healthy individuals and physically conditioned subjects. A sinus rate of <40 beats/min in the awake state in the absence of physical conditioning generally is considered abnormal. Sinus pauses and sinus arrest result from failure of the SA node to discharge, producing a pause without P waves visible on the ECG (Fig. 15-3). Sinus pauses of up to 3 s are common in awake athletes, and pauses of this duration or longer may be observed in asymptomatic elderly subjects. Intermittent failure of conduction from the SA node produces sinus exit block. The severity of sinus exit block may vary in a manner similar to that of AV block (see later). Prolongation of conduction from the sinus node will not be apparent on the ECG; second-degree SA block will produce intermittent conduction from the SA node and a regularly irregular atrial rhythm.
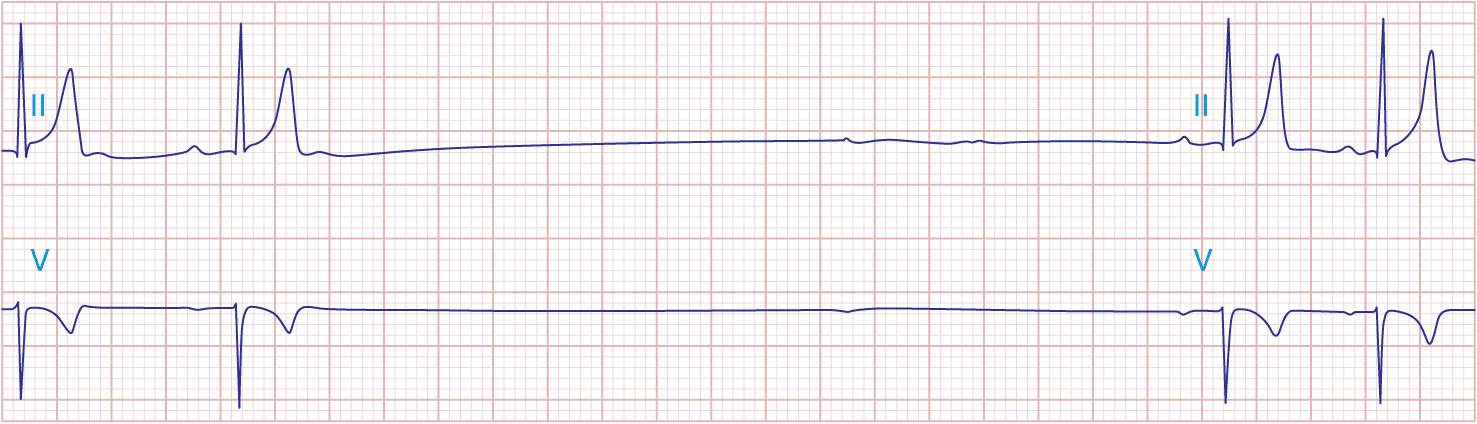
FIGURE 15-3
Sinus slowing and pauses on the ECG. The ECG is recorded during sleep in a young patient without heart disease. The heart rate before the pause is slow, and the PR interval is prolonged, consistent with an increase in vagal tone. The P waves have a morphology consistent with sinus rhythm. The recording is from a two-lead telemetry system in which the tracing labeled II mimics frontal lead II and V represents Modified Central Lead 1, which mimics lead V1 of the standard 12-lead ECG.
Type I second-degree SA block results from progressive prolongation of SA node conduction with intermittent failure of the impulses originating in the sinus node to conduct to the surrounding atrial tissue. Second-degree SA block appears on the ECG as an intermittent absence of P waves (Fig. 15-4). In type II second-degree SA block, there is no change in SA node conduction before the pause. Complete or third-degree SA block results in no P waves on the ECG. Tachycardia-bradycardia syndrome is manifest as alternating sinus bradycardia and atrial tachyarrhythmias. Although atrial tachycardia, atrial flutter, and atrial fibrillation may be observed, the latter is the most common tachycardia. Chronotropic incompetence is the inability to increase the heart rate in response to exercise or other stress appropriately and is defined in greater detail later.
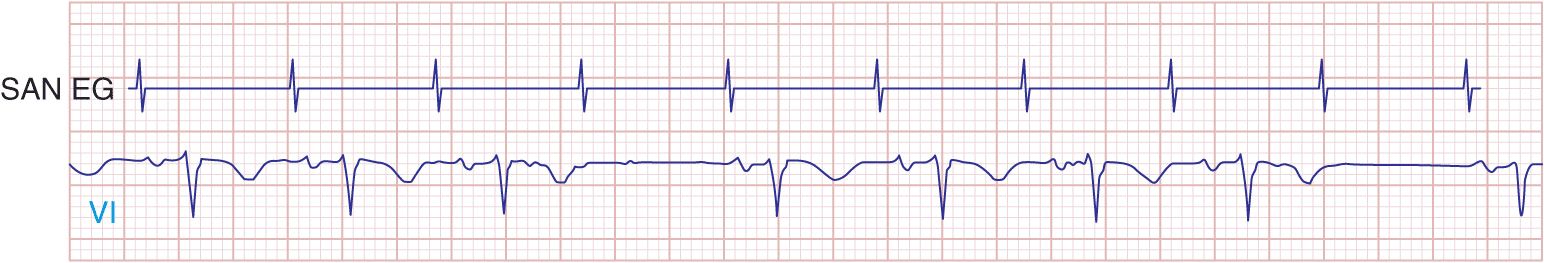
FIGURE 15-4
Mobitz type I SA nodal exit block. A theoretical SA node electrogram (SAN EG) is shown. Note that there is grouped beating producing a regularly irregular heart rhythm. The SA node EG rate is constant with progressive delay in exit from the node and activation of the atria, inscribing the P wave. This produces subtly decreasing P-P intervals before the pause, and the pause is less than twice the cycle length of the last sinus interval.
Diagnostic testing
SA node dysfunction is most commonly a clinical or electrocardiographic diagnosis. Sinus bradycardia or pauses on the resting ECG are rarely sufficient to diagnose SA node disease, and longer-term recording and symptom correlation generally are required. Symptoms in the absence of sinus bradyarrhythmias may be sufficient to exclude a diagnosis of SA node dysfunction.
Electrocardiographic recording plays a central role in the diagnosis and management of SA node dysfunction. Despite the limitations of the resting ECG, longer-term recording employing Holter or event monitors may permit correlation of symptoms with the cardiac rhythm. Many contemporary event monitors may be automatically triggered to record the ECG when certain programmed heart rate criteria are met. Implantable ECG monitors permit long-term recording (12–18 months) in particularly challenging patients.
Failure to increase the heart rate with exercise is referred to as chronotropic incompetence. This is alternatively defined as failure to reach 85% of predicted maximal heart rate at peak exercise or failure to achieve a heart rate >100 beats/min with exercise or a maximal heart rate with exercise less than two standard deviations below that of an age-matched control population. Exercise testing may be useful in discriminating chronotropic incompetence from resting bradycardia and may aid in the identification of the mechanism of exercise intolerance.
Autonomic nervous system testing is useful in diagnosing carotid sinus hypersensitivity; pauses >3 s are consistent with the diagnosis but may be present in asymptomatic elderly subjects. Determining the intrinsic heart rate (IHR) may distinguish SA node dysfunction from slow heart rates that result from high vagal tone. The normal IHR after administration of 0.2 mg/kg propranolol and 0.04 mg/kg atropine is 117.2 – (0.53 × age) in beats/min; a low IHR is indicative of SA disease.
Electrophysiologic testing may play a role in the assessment of patients with presumed SA node dysfunction and in the evaluation of syncope, particularly in the setting of structural heart disease. In this circumstance, electrophysiologic testing is used to rule out more malignant etiologies of syncope, such as ventricular tachyarrhythmias and AV conduction block. There are several ways to assess SA node function invasively. They include the sinus node recovery time (SNRT), defined as the longest pause after cessation of overdrive pacing of the right atrium near the SA node (normal: <1500 ms or, corrected for sinus cycle length, <550 ms), and the sinoatrial conduction time (SACT), defined as one-half the difference between the intrinsic sinus cycle length and a noncompensatory pause after a premature atrial stimulus (normal <125 ms). The combination of an abnormal SNRT, an abnormal SACT, and a low IHR is a sensitive and specific indicator of intrinsic SA node disease.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


