Definition of Sudden Cardiac Death
Sudden cardiac death (SCD) describes the unexpected natural death due to a cardiac cause within a short time period from the onset of symptoms in a person without any prior condition that would appear imminently fatal. It is most often due to a sustained ventricular tachyarrhythmia. Although many cardiovascular disorders increase the risk of SCD, the presence or absence of preexisting cardiovascular disease is not necessary.1 Prodromal symptoms such as palpitations, chest pain, and dyspnea may suggest a cardiovascular etiology such as arrhythmia, ischemia, or congestive heart failure but are not specific. The definition of SCD includes the time interval from onset of the symptoms leading to collapse and then to death, the unexpected nature of the event, and the specific cause of death. Recent definitions have focused on time intervals of 1 hour or less, which normally identify SCD populations having a 90% or more proportion of arrhythmic death.2,3 However, because 80% of sudden deaths occur in the home environment and up to 40% are unwitnessed, the interval between symptoms onset and death is frequently unknown, rendering precise measurement of SCD rates difficult.4,5
Epidemiology
SCD accounts for more than 460,000 deaths yearly in the United States, with exact totals depending on the definition used.1,6 When the definition of SCD is restricted to death less than 2 hours from onset of symptoms, 12% of all natural deaths are sudden and 88% of these are due to cardiac disease. In autopsy-based studies, a cardiac etiology of sudden death has been reported in 60% to 70% of sudden death victims.5
The US Centers for Disease Control and Prevention has estimated the SCD incidence per 1000 persons in 1999 to be 2.1 in men and 1.4 in women, with SCD accounting for 63% of all deaths attributed to cardiac disease.6 The average age of cardiac arrest victims is approximately 65 years, and 70% to 80% are men.7 In the United States, several population-based studies have documented an age-adjusted decline in SCD rates of more than 8% over the last 15 years.1 SCD rates in other developed countries are comparable to those in the United States: The World Health Organization reported an annual incidence of SCD of 1.9 per 1000 persons in men and 0.6 in women.8 SCD rates in developing countries are considerably lower, paralleling the rates of ischemic heart disease, the most common substrate for SCD in developed nations (Fig. 49–1).
Figure 49–1
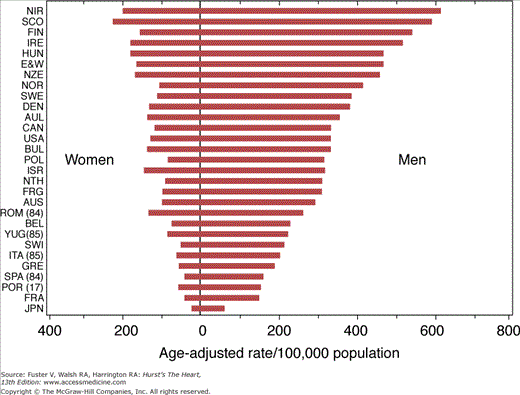
Sudden cardiac death (SCD) rates by sex and country, ages 35 to 74 years, compiled from death certificates by the World Health Organization, Geneva, 1986. Reproduced with permission from Manolio TA, Furberg CD. Epidemiology of SCD. In: Akhtar M, Myerburg RJ, Ruskin JN, eds. SCD. Baltimore, MD: Williams & Wilkins; 1994:3.
The incidence of SCD increases along with the prevalence of ischemic heart disease at older ages (Fig. 49–2).9 Among sudden natural deaths, the proportion with cardiac causes also increases with advancing age. Among patients with coronary heart disease (CHD), however, the proportion of cardiac deaths that are sudden decreases from more than 74% of deaths in those aged 35 to 44 years to <60% of those aged 75 to 84 years.1,9
Figure 49–2
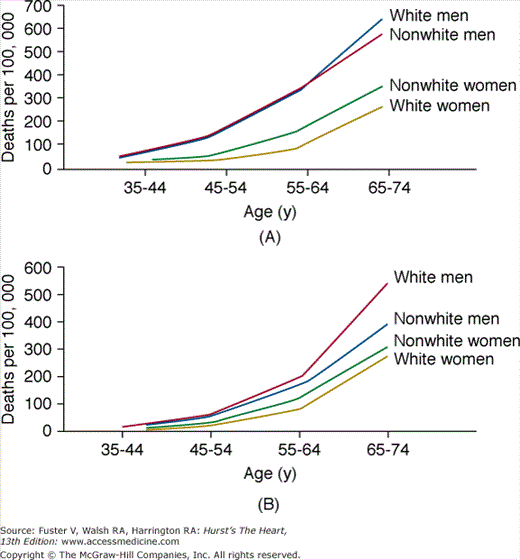
Plots of mortality rates (deaths per 100,000) for ischemic heart disease occurring (A) out of hospital or in emergency room (an estimate for sudden cardiac death rate) and (B) occurring in the hospital, by age, sex, and race in 40 states during 1985. From the National Center for Health Statistics. Reproduced with permission from Gillum RF. Sudden coronary death in the United States: 1980-1985. Circulation. 1989;79:756-765.
SCD accounts for approximately 20% of all sudden deaths in patients younger than age 20.10 Structural cardiac abnormalities can be identified in the majority of young victims of SCD11; however, recent autopsy-based studies suggest that 20% to 35% of sudden deaths in young adults occur in the absence of identifiable structural abnormalities.12,13 Many of these are likely due to genetically based arrhythmogenic disorders. These topics are discussed in further detail below (also see Chap. 36).
The annual incidence of SCD has been shown to be higher in African Americans than whites in numerous studies.1,14 An analysis of cardiac death rates in the United States between 1989 and 1998 showed that the age-adjusted rates of SCD per 1000 persons were 5.0 for black men, 4.1 for white men, and 2.1 for Asians. Likewise, in a recent analysis of sudden deaths in young female military recruits, African American women were found to have a risk ratio of 10.2 compared with other races.15 The reasons for these findings are unclear. Postmortem analyses show that differences in rates of sudden death may be due to an increased prevalence of hypertension, left ventricular hypertrophy, diabetes, and tobacco use.16 Other factors that must be considered include limitations in access to preventive care,17 prehospital delays in patient activation of emergency medical services, and disparities in health system quality that disproportionately affect minorities.18 These issues warrant further investigation.
Age-adjusted SCD rates are much higher in men than in women, reflecting sex-related differences in the incidence of CHD.1,9 As is the case with coronary disease, however, this disparity decreases with advancing age, with a male: female ratio for SCD of 7:1 in 45- to 64-year-olds but only 2:1 in 65- to 74-year-olds.
Women are more likely than men (64% vs 50%) to suffer SCD without prior evidence of coronary heart disease.9 Among survivors of cardiac arrest, women are more likely than men to have other forms of structural heart disease (valvular heart disease, dilated cardiomyopathy) or a structurally normal heart.19 SCDs in women are much more likely to occur outside the hospital than in men, and the aforementioned decline in sudden death rates has been noted to be smaller in women (6%) than men (12%). In fact, the rate of sudden death has increased by 21% in women aged 35 to 44 years. The reasons for these findings are myriad and may include less aggressive treatment,17,20 patients’ lack of awareness of the importance of cardiovascular signs and symptoms, and the greater likelihood of atypical cardiac symptoms in women.
Only a fraction of patients survive a cardiac arrest, and there has been considerable interest in identifying populations at risk for sustained ventricular arrhythmias. More that 80% of SCDs occur in patients with underlying coronary disease. The risk factors for SCD therefore largely reflect those for CHD, with hypertension, hypercholesterolemia, smoking, and diabetes appearing as significant risk factors in multiple studies (Table 49–1). The majority of SCDs occur in persons with multiple risk factors (Fig. 49–3).
| Study | Study Population | Risk Factors for SCD |
|---|---|---|
| Kannel et al9,21 (Framingham Study) | 5128 men and women, age 30-62, no CHD at entry: 546 CHD deaths over 26 years, 46% (men) and 35% (women) SCD | Men
Women
|
| Hinkle et al22 | 269,755 men, age 20-65: 1839 CHD deaths over 5 y, 60% SCD | Hypertension Cigarette smoking Alcohol History of CHD LVH (by ECG) Enlarged heart (CXR) CHF PVCs |
| Wannamethee et al273 | 7735 British men, age 40-59: 488 CHD events, 117 SCD over 8 y | History of CHD Physical inactivity Systolic blood pressure Cholesterol Cigarette smoking Arrhythmia Resting hear rate >90 Alcohol Diabetes mellitus |
| Albert et al274 | 121,701 women age 30-55: 1110 CV deaths, 244 SCD over 12 y | Hypertension Cigarette smoking Family history CHD Obesity |
Figure 49–3
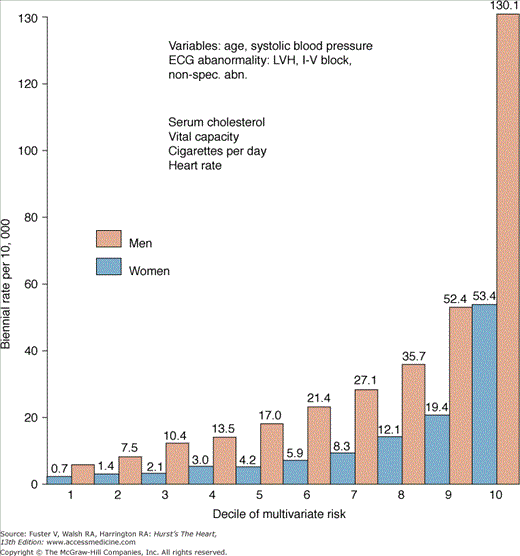
Risk of sudden cardiac death by decile of multivariate risk: 26-year follow-up, the Framingham Study. ECG, electrocardiographic; I-V, intraventricular; LVH, left ventricular hypertrophy; non-spec. abn., nonspecific abnormality. Reprinted with permission from Kannel WB, Schatzkin A. Sudden death: lessons from subsets in population studies. J Am Coll Cardiol. 1985;5(suppl):141B-149B.
Despite the fact that studies have shown a strong relationship between risk factors for CHD and SCD, no clear modifiable risk factors that are specific for SCD have been identified once CHD is established.21,22 Rather, the likelihood of SCD once CHD is established is most strongly related to the degree of cardiac damage, as reflected by structural and electrocardiographic abnormalities and the severity of clinical heart failure.
The inability to determine epidemiological risk factors specific for SCD reflects the fact that these factors are linked to chronic disease processes that create the structural basis for sustained arrhythmia. These structural abnormalities may be necessary but are not sufficient to cause an episode of SCD. In the future, genotypic and other new biomarkers may improve the stratification of risk for sudden death. Novel markers to assess risk of sudden death are currently under investigation. Hemostatic factors,23 inflammatory markers, and polymorphisms in cardiac β-receptors24,25 show promise.
Observations suggest that lifestyle factors such as alcohol consumption, cigarette smoking, exercise, and stress are significantly correlated with the risk of dying suddenly.21,26,27 Individuals who consume large amounts of alcohol (more than 5 drinks per day) have increased risks of ventricular arrhythmia and SCD.28 However, a prospective analysis of 21,537 subjects in the Physicians’ Health Study demonstrated a decreased risk of SCD in men who consumed light-to-moderate amounts of alcohol (2 to 6 drinks per week) compared with those who drank rarely or never.26
Cigarette smoking is one of the few coronary risk factors associated with a disproportionate increase in the risk of SCD. In the Framingham Study, the annual incidence of SCD increased from 13 per 1000 in nonsmokers to 31 per 1000 in those smoking more than 20 cigarettes per day.29 Smoking has been shown to induce physiologic changes that predispose to SCD, such as increased platelet adhesiveness and catecholamine release, decreased ventricular fibrillation threshold, acceleration of heart rate, increased blood pressure, coronary spasm, reduced oxygen-carrying capacity by accumulation of carboxyhemoglobin, and impairment of myoglobin utilization.29 A postmortem study linked smoking and the presence of acute coronary thrombus. Fresh thrombus was found in more than 50% of men who died suddenly, and cigarette smoking was a risk factor in 75% of these men, compared with 41% of the men with stable plaques.30 Those who stop smoking have a prompt reduction in CHD mortality rate irrespective of the duration of previous smoking.31 In addition, recent studies have documented significant reductions in acute myocardial infarction rates after the institution of public smoking bans,32 although specific effects on SCD rates have not yet been explored.
There are many reports linking stress, particularly emotional stress, to SCD. Patients experiencing intense anger tend to have higher rates of implantable cardioverter-defibrillator (ICD) discharge,27 and an estimated seven times greater relative risk of appropriate discharge occurs during mental and physical stress.33 ICD discharge rates in New York City remained elevated for up to 30 days after the 2001 terrorist attacks.34
There is increasing evidence that regular physical activity may help prevent CHD and its complications; however, the benefits of vigorous exercise in patients with known CHD is controversial. Several clinical and autopsy-based studies have reported exercise as a trigger for SCD.35,36 Emergency medical records have shown that 11% to 17% of adults with CHD and SCD collapse during or immediately after exertion, but the amount of exertion is rarely quantified.37 Cardiac arrest rates of 1 in 12,000 to 15,000 in cardiac rehabilitation programs and 1 per 2000 during cardiac stress testing have been reported.38 These rates are at least six times higher than those for patients not known to have heart disease.37
Although the absolute risk of sudden death during any particular episode of vigorous exertion is low (1 sudden death per 1.51 million episodes of exertion), the Physicians’ Health Study estimated that the risk of sudden death increases more than 16-fold in the first 30 minutes of vigorous activity.36 Nevertheless, there is ample experimental evidence that regular exercise may prevent ischemia-induced ventricular fibrillation and death,39 and habitual vigorous exercise attenuates the relative risk of sudden death associated with vigorous exertion.36 It therefore appears that regular participation in moderate-intensity activities is associated with reduced rates of cardiovascular morbidity and mortality, but the risks of SCD and myocardial infarction are transiently increased during acute bouts of high-intensity activity.
SCD in competitive athletes is rare (see Chap. 101). Unfortunately, SCD is often the first manifestation of the underlying cardiac disease that is present in the majority of these patients.40 Age has been shown to be the most useful variable in predicting the type of underlying cardiac disease (Fig. 49–4). In athletes younger than 35 years of age, the majority of SCDs arise from a variety of congenital cardiovascular diseases, most commonly hypertrophic cardiomyopathy and congenital coronary artery anomalies.41 Less common structural conditions include myocarditis, aortic dissection (eg, related to Marfan syndrome), aortic stenosis, dilated cardiomyopathy, and arrhythmias associated with mitral valve prolapse. Other disorders such as arrhythmogenic right ventricular cardiomyopathy (ARVC) may be more frequent in endemic areas.42 SCD among young athletes without structurally identifiable cardiac abnormalities is reportedly rare,40 but was recently found to be much more common among young active-duty military members.13 These cases are likely to be related to inherited arrhythmia disorders such as long-QT syndrome, Wolff-Parkinson-White syndrome, and others (see Chap. 36 and below). Coronary artery disease is an uncommon cause of SCD in athletes younger than 35 years.43
Figure 49–4
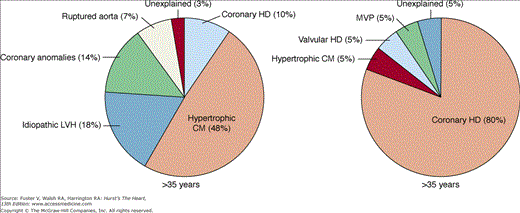
Causes of sudden cardiac death in competitive athletes by age group. There is evidence for structural heart disease in nearly all athletes who die suddenly of cardiac causes. In athletes younger than 35 years, hypertrophic cardiomyopathy is more prevalent, whereas in those older than 35 years, coronary heart disease is the most frequent cause. CM, cardiomyopathy; HD, heart disease; LVH, left ventricular hypertrophy; MVP, mitral valve prolapse. Reproduced with permission from Maron BJ, Epstein SE, Roberts WC. Causes of sudden death in competitive athletes. J Am Coll Cardiol. 1986;7:204-214.
Because of the dramatic and often public nature of SCD in athletes and the medical-legal climate, the need for extensive preparticipation screening of athletes for cardiac conditions has been debated. US guidelines for such screening have been published and they focus on detailed personal and family history and physical examination.44 A long-standing screening program in Italy has credited the use of routine electrocardiography with identifying cardiomyopathies and significantly reducing the rate of SCD during athletic activities,45,46 but the necessity of routine electrocardiography has been questioned.47 Both US and European guidelines have also been published outlining which athletes with cardiac arrhythmias can participate in competitive athletics48,49 (see Chap. 101).
Blunt, nonpenetrating, and usually innocent-appearing chest blows leading to ventricular arrhythmia and sudden death have also been reported as causes of SCD in young persons.50 Termed commotio cordis, these events often occur during organized sports but also occur in other settings. Most deaths occur with low energy and velocity projectiles striking the chest.
Mechanism of SCD
A vast majority of patients who have experienced SCD have cardiac structural abnormalities. In the adult population, these consist predominantly of CHD, cardiomyopathies, valvular heart disease, and abnormalities of the conduction system. These structural changes provide the substrate for ventricular tachyarrhythmias, the cause of SCD in most cases. It is important to recognize the role of triggering factors such as ischemia, hemodynamic changes, fluctuations in the autonomic nervous system, electrolyte abnormalities, and proarrhythmic effects of drugs in the initiation of ventricular arrhythmias resulting in SCD (Fig. 49–5).51,52 Strategies aimed at eliminating or reducing the triggers of arrhythmias may prove to be effective short- and midterm solutions because most structural abnormalities, once present, cannot be cured.
Figure 49–5
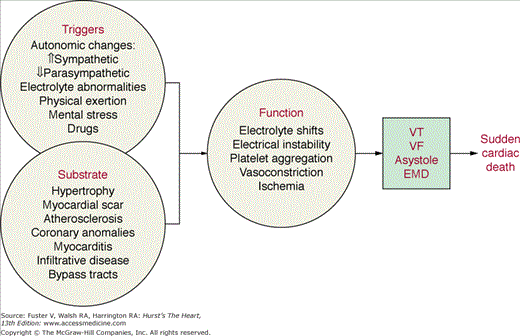
Interaction between structural cardiac abnormalities, functional changes, and triggering factors in the pathophysiology of sudden cardiac death. The role of triggering factors, such as changes in autonomic tone or reflexes, is increasingly being recognized. EMD, electromechanical dissociation; VF, ventricular fibrillation; VT, ventricular tachycardia.
Ventricular fibrillation is the first recorded rhythm in approximately 75% of patients who have cardiac arrest.53 Sustained ventricular tachycardia is only rarely (<2%) documented as the initial rhythm, but it is unknown how often it precedes and precipitates ventricular fibrillation. In a series of 157 ambulatory patients who were wearing an ECG monitor at the time of their cardiac arrest, primary ventricular fibrillation was documented in 8%, ventricular tachycardia degenerating into ventricular fibrillation in 62%, and torsade de pointes in 13%.54 Likewise, in patients with implantable cardioverter defibrillators, 90% of appropriate arrhythmia detections are for ventricular tachycardia (VT) rather than ventricular fibrillation (VF).55
Electromechanical dissociation and asystole are found in approximately 30% of patients experiencing cardiac arrest, and this finding is usually related to the time interval from collapse to first monitoring of the rhythm, suggesting that it is often a later manifestation of cardiac arrest.53 The incidence of bradycardia as the first documented rhythm varies according to the population studied. In patients who have died suddenly while wearing an ambulatory ECG monitor, bradyarrhythmias as the initial rhythm were documented infrequently, and ventricular tachyarrhythmias were most often the mode of cardiac arrest, even in patients with preexisting atrioventricular or intraventricular conduction defects.54 In a small group of patients with severe congestive heart failure awaiting cardiac transplant, bradycardia or electromechanical dissociation at the time of death was more frequent than ventricular arrhythmia.56 We believe that in this population, bradycardia reflects the unrelenting failure of the severely impaired heart and is not a primary cause of sudden death unless the bradyarrhythmia allows for the development of a tachyarrhythmia. An understanding of the mechanisms responsible for ventricular arrhythmia is essential in its prevention and treatment (see Chaps. 38 and 42).
Acute myocardial ischemia leads to intracellular and extracellular acidosis and loss of myocellular membrane integrity with efflux of potassium and influx of calcium. These biochemical abnormalities have electrophysiological consequences, including decreases in the amplitude and upstroke velocity of the cardiac action potential, inhomogeneous depolarization of the resting membrane potential, and shortening of action potential duration.57 Fast sodium and slow calcium channels in partially depolarized fibers may remain inactive, thereby prolonging refractoriness even after completion of repolarization. This may further contribute to electrical inhomogeneities within and around the ischemic zone, causing conduction delays, unidirectional block, and reentrant arrhythmias.58
Ventricular arrhythmias during experimental coronary occlusion occur in two peaks, the first between 2 and 8 minutes (phase IA) and the second at 15 to 45 minutes (phase IB). Rapid polymorphic VT and VF are the characteristic arrhythmias during the early stages of ischemia.59 Activation mapping during VF has demonstrated that the initial arrhythmias are due to reentry, which is facilitated by the inhomogeneous conduction velocities and refractory periods in and around the ischemic zone. The second peak of ventricular arrhythmias coincides with a peak in catecholamine release. Automatic and triggered rhythms have been implicated in these arrhythmias. Reentrant rhythms may also occur, attributed to cellular uncoupling related to impaired function of gap junctions.60
Within the first 3 days of myocardial infarction, SCD may occur due to ventricular fibrillation initiated by early, frequent premature ventricular complexes (PVCs). Such PVCs have been shown in experimental models to be predominantly due to impulse formation, consistent with abnormal automaticity. Other manifestations of abnormal automaticity are the accelerated idioventricular rhythms signifying reperfusion. These arrhythmias appear to arise, for the most part, from surviving Purkinje fibers in the subendocardial border zone of a transmural infarction. They have no prognostic significance for development of late arrhythmias and usually subside after 2 to 3 days at approximately the same time that the resting membrane potential and action potential duration of Purkinje fibers normalize.57
In the late phases after myocardial infarction, when the infarction is healed, reentrant excitation is the principal mechanism of ventricular arrhythmias. Critical areas of the reentrant circuit are formed by surviving myocardial cells in the epicardial and endocardial border zone of a healed infarction, as well as surviving intramural fibers within the infarct zone.61 It appears that the rate, time, and degree of myocardial reperfusion influence the incidence, rate, and duration of these arrhythmias. More work is needed to further define these relationships (see Chap. 38).
Left ventricular dysfunction has been identified as the strongest independent predictor of SCD.62 Despite the clinical recognition that acute heart failure can precipitate ventricular tachyarrhythmias, the mechanism by which this occurs is incompletely understood. Besides mechanisms related to acute and chronic ischemia, it has been shown that acute changes in the mechanical state of the heart related to altered preload and contractility can have direct electrophysiologic effects that may precipitate arrhythmias; this relationship is referred to as mechanoelectrical feedback. An increase in right ventricular pressure has been shown to shorten action potential duration in humans.63 An increase in both left ventricular preload and contractility has been shown to shorten action potential duration and refractoriness in the canine ventricle as well.64 There is some evidence that these changes may be mediated through the β-adrenergic receptor. Downstream effects leading to fluctuation of intracellular calcium levels or to an increase in a cyclic AMP-mediated potassium current may serve as the cellular mechanism by which this phenomenon occurs.64 Recent mechanistic evidence has also pointed to calcium handling in the sarcoplasmic reticulum, mediated by defective regulation of the cardiac ryanodine receptor.65
Cardiac Diseases Associated with SCD
| Ischemic heart disease | |
| Coronary atherosclerosis | Coronary artery spasm |
| Acute myocardial infarction | Coronary artery dissection |
| Chronic ischemic cardiomyopathy | Coronary arteritis |
| Anomalous origin of coronary arteries | Small vessel disease |
| Hypoplastic coronary artery | |
| Nonischemic heart disease | |
| Cardiomyopathies | Drug-induced and other toxic agents |
| Idiopathic dilated cardiomyopathy | Antiarrhythmic drugs (classes Ia, Ic, and III) |
| Hypertrophic cardiomyopathy | Erythromycin, clarithromycin |
| Hypertensive cardiomyopathy | Methadone |
| Arrhythmogenic right ventricular cardiomyopathy | Astemizole |
| Left ventricular noncompaction | Terfenadine |
| Infiltrative and inflammatory heart disease | Pentamidine |
| Sarcoidosis | Ketoconazole |
| Amyloidosis | Trimethoprim-sulfamethoxazole |
| Hemochromatosis | Psychotropic drugs (tricyclic antidepressants, haloperidol, phenothiazines) |
| Myocarditis | Probucol |
| Valvular heart disease | Cisapride |
| Aortic stenosis | Cocaine |
| Aortic regurgitation | Chloroquine |
| Mitral valve prolapse | Alcohol |
| Infective endocarditis | Phosphodiesterase inhibitors |
| Congenital heart disease | Organophosphates |
| Tetralogy of Fallot | Electrolyte abnormalities |
| Transposition of the great vessels (post–Mustard-Senning) | Hypokalemia |
| Ebstein anomaly | Hypomagnesemia |
| Pulmonary vascular obstructive disease | Hypocalcemia |
| Congenital aortic stenosis | Anorexia nervosa and bulimia |
| Primary electrical abnormalities | Liquid protein dieting |
| Long-QT syndrome | Diuretics |
| Short-QT syndrome | |
| Wolff-Parkinson-White syndrome | |
| Congenital AV block | |
| Brugada syndrome | |
| Catecholaminergic polymorphic VT | |
| Idiopathic ventricular fibrillation | |
| Early repolarization variants |
In survivors of cardiac arrest, CHD is present in 40% to 86% of patients, depending on the age and sex of the population.66 Patients with known CHD accounted for 20% to 34% of SCDs in the Framingham cohort.9 SCD was the first symptom of CHD in approximately 10% of all coronary events.
Although many who suffer SCD have severe multivessel coronary disease, fewer than half of the patients resuscitated from VF evolve evidence of myocardial infarction by elevated cardiac biomarkers, and less than 20% have transmural (Q wave) myocardial infarction.53 Holter monitoring at the time of arrest has infrequently shown evidence of ischemic ECG changes before the event.51,67 In postmortem examinations and in catheterization studies, there was a significant (75%-85%) stenosis in at least two major coronary arteries in as many as 76% of patients. Acute coronary thrombosis has been reported in 25% to 48% of survivors of out-of-hospital cardiac arrest undergoing immediate coronary angiography.66,68 Detailed pathologic studies have confirmed the presence of acute coronary arterial lesions (plaque fissure, plaque hemorrhage, and thrombosis) in up to 95% of patients dying suddenly, but only a fraction had total occlusion.30,69
Coronary collateralization may play an important role in the presentation of coronary artery disease as SCD. It has been hypothesized that chronic ischemia may be a stimulus for development of coronary collaterals, which in turn could have a protective effect during acute coronary occlusion. The mitigating effect of coronary collateralization is supported by a study of exercise testing in 894 healthy men followed-up for a mean of 12.7 years. In this study, the initial coronary event was acute myocardial infarction or SCD in 73% of those with a normal stress test result, as opposed to 20% of those with an abnormal stress test result.70 These findings might also be related to ischemic preconditioning, which is associated with reductions in VF in animal models.71
Several nonatherosclerotic diseases of the coronary arteries are associated with increased risk of SCD precipitated by cardiac ischemia. Congenital coronary artery anomalies, found in approximately 1% of all patients undergoing angiography and in 0.3% of patients undergoing autopsy, have been associated with SCD, often exercise-related, in up to approximately 17% of competitive athletes with confirmed cardiovascular sudden death.41 Origin of the left main coronary artery from the right aortic sinus or origin of the right coronary artery from the left coronary sinus are the variants most frequently associated with SCD.
Life-threatening ventricular arrhythmias and SCD have been described in patients with coronary artery spasm (Prinzmetal or variant angina). Significant arrhythmias during attacks of variant angina have been documented in these patients.72 Calcium channel blockers are effective in many patients in preventing coronary spasm and also appear to protect from malignant ventricular arrhythmias if the attacks can be completely abolished. Predictors of major coronary events in this population include systemic hypertension, luminal irregularities on coronary arteriography, and QT dispersion.72
SCD has been described as a rare complication of coronary artery dissection in Marfan syndrome, during the puerperium, secondary to trauma or coronary catheterization, as a consequence of syphilitic aortitis, or as an extension of aortic dissection. Myocardial bridges have been reported in association with SCD during exercise, but they are also an incidental finding at autopsy in up to 25% of patients dying of other causes.73 Coronary arteritis and subsequent infarction have been reported in Kawasaki disease, giant-cell arteritis, Beçhet disease, systemic lupus erythematosis, and Churg-Strauss syndrome. Coronary occlusion can also occur as a complication of infective endocarditis, particularly involving the aortic valve (see Chap. 55).
Idiopathic dilated cardiomyopathy is the substrate for approximately 10% of SCDs in the adult population. The mortality rate for idiopathic dilated cardiomyopathy is high, reaching 10% to 50% annually, and is most closely tied to the severity of pump dysfunction.74 Mortality rates are higher among patients with advanced heart failure, but the proportion of sudden to nonsudden cardiac death is not increased.75 In an overview of 14 studies including 1432 patients with idiopathic dilated cardiomyopathy, the mean mortality rate after a follow-up of 4 years was 42%, with 28% of deaths classified as sudden.74 SCD in idiopathic dilated cardiomyopathy is usually attributed to both polymorphic and monomorphic ventricular tachyarrhythmias occurring in the setting of a high frequency of complex ventricular ectopy.76 However, the terminal event may also be due to pump failure with asystole or electromechanical dissociation, especially in patients with advanced left ventricular dysfunction.56
Risk stratification of patients with idiopathic dilated cardiomyopathy is difficult because there are few clinical predictors specific for SCD. The only clinical variable that identifies patients with a higher risk of SCD is unexplained syncope, and these patients should undergo further evaluation. The prognostic value of simple and complex PVCs as well as nonsustained ventricular tachycardia (NSVT) is limited by the extremely high prevalence of these arrhythmias in patients with idiopathic dilated cardiomyopathy.76 The induction of polymorphic VT or VF during electrophysiologic (EP) testing is nonspecific, and the probability of inducing sustained monomorphic VT in this population is less than 10%.77 Therefore, EP study is not considered a useful screening tool in patients with nonischemic cardiomyopathy. However, when sustained monomorphic VT occurs in patients with dilated cardiomyopathy, the mechanism is usually reentrant and potentially amenable to catheter ablation.78 (see Chaps. 32 and 44).
Hypertrophic cardiomyopathy (HCM), caused by a variety of mutations in genes encoding cardiac sarcomere proteins, is classically associated with asymmetric septal hypertrophy and myofibrillar disarray, but has diverse clinical presentations. The incidence of SCD in selected families with HCM treated at large referral centers has been reported as high as 2% to 4% per year in adults and 4% to 6% per year in children and adolescents,79 but is probably closer to 1% per year in unselected populations.80,81
A number of factors have been studied to aid in distinguishing the minority of HCM patients at high risk for SCD from the majority of patients at much lower risk. The highest risk features include a clinical history of spontaneous ventricular arrhythmias, sudden death in family members, onset of symptoms in childhood, and syncope or near-syncope, especially when exertional or recurrent and unrelated to neurocardiogenic mechanisms.82 Other risk factors with a lower predictive value include extreme left ventricular wall thickness (≥3.0 cm),83 hypotensive response to exercise,84 unsustained VT during ambulatory monitoring (particularly repetitive or prolonged bursts), and significant (>30-50 mm Hg) resting left ventricular outflow tract gradient. Although none of these factors alone have a high positive predictive value, they do have high negative predictive value, particularly in combination.81 Current consensus guidelines recommend ICD implantation for patients with ≥1 major risk factors for SCD (class IIa),85 and ICDs appear to be effective at aborting ventricular arrhythmias in these patients.86
Because of the clinical heterogeneity of HCM, there is hope that genotypic analysis will provide further guidance on SCD risk. For example, some mutations of the β-myosin heavy chain appear to be associated with a benign prognosis, whereas mutations in cardiac troponin T have been associated with a mild degree of hypertrophy but a high incidence of SCD in selected kindreds.87,88 As with most genetic disorders, however, the phenotypic expression of the same genetic abnormality is highly variable in HCM, and clinical decisions cannot be reliably made at this time based solely on genotype89 (see Chaps. 33 and 82).
Left ventricular hypertrophy (LVH) has been identified as one of the strongest independent risk factors for acute myocardial infarction, congestive heart failure, and sudden death.90,91 In the Framingham Study, ECG evidence of LVH doubled the risk of SCD. Echocardiographic studies showed an incremental risk for cardiovascular deaths of 1.73 in men and 2.12 in women for each 50-g increment in the left ventricular mass index.92 Decreased coronary blood flow, decreased flow reserve, and endothelial dysfunction may all be factors favoring the development of transient ischemia in the setting of LVH, and long-term, repeated transient ischemic episodes can lead to interstitial fibrosis, which may underlie the arrhythmias in this population.93 Another potential contributing factor to the increased risk of SCD in hypertensive cardiomyopathy are electrolyte disturbances associated with diuretic therapy.94
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is characterized by fatty or fibrofatty replacement of myocardium, primarily in the right ventricle (RV), and ventricular tachyarrhythmias, usually of RV origin. It is a rare cause of SCD except in a few endemic regions and is a familial disorder in approximately 30% of cases.
Multiple genetic mutations have now been reported as causing ARVC. The first reported mutation involves the cardiac ryanodine receptor (RyR2), the major calcium release channel on the sarcoplasmic reticulum in cardiomyocytes.95 More recently, mutations have been reported from genes encoding four related proteins (plakoglobin,96 desmoplakin,97 plakophilin-2,98 and desmoglein-299) associated with desmosomes, which anchor intermediate filaments to the cell membrane in adjoining cells. An autosomal-recessive mutation of the plakoglobin gene causes Naxos disease, characterized by ARVC and skin and hair abnormalities.96 Desmosomal proteins are now thought to be involved in at least 40% of ARVC cases, but mutations of unrelated proteins, including transforming growth factor-β3,100 have also been reported.
Recurrent VTs with multiple left bundle-branch block morphologies typifies ARVC. Patchy myocarditis, programmed cell death, and/or congenital abnormalities of development appear to lead to myocardial atrophy and repair by fibrofatty replacement, which forms the basis for ventricular reentry. The left ventricle and ventricular septum can be involved, especially later in the course of the disease, and such involvement confers a poor prognosis.101
The electrocardiographic manifestations of ARVC in sinus rhythm include T wave inversion in V1 to V3 or complete or incomplete right bundle-branch block. Intraventricular conduction delay may produce a terminal notch on the QRS complex called an epsilon wave in approximately 50% of patients. Ventricular ectopy is usually of a left bundle-branch pattern, typically with a more leftward axis than the usually benign ectopy that arises in the RV outflow tract.
The diagnosis of ARVC is most easily made by magnetic resonance imaging; consensus diagnostic criteria were published in 1994.102 Immunohistochemical analysis of endomyocardial biopsy specimens revealing diffusely reduced plakoglobin signal at intercalated discs has also recently been found to be both sensitive and specific for ARVC.103
The course and prognosis of ARVC are highly variable and difficult to predict even in patients with overt disease and significant ventricular arrhythmias. ICDs are the primary therapy in those with sustained VT with hemodynamic compromise or VF, and nonrandomized studies have suggested improved survival in patients with serious symptoms and high-risk mutations.104,105 Sustained, tolerated VT can be treated successfully with catheter ablation,106 but recurrent VT is common,107 and because ARVC tends to be a progressive condition, these patients are usually treated with ICDs as well. More data are required to define which asymptomatic patients with ARVC benefit from this therapy (see Chap. 82).
The risk of SCD in asymptomatic patients with aortic stenosis or regurgitation appears to be low.108,109 In contrast, there appears to be an increased risk of SCD after aortic valve replacement for aortic stenosis or regurgitation. In 831 patients receiving a Bjork-Shiley prosthesis in the aortic (341 patients), mitral (345 patients), or double-valve (145 patients) position, the incidence of SCD in the subgroups was 1.8%, 3.5%, and 4%, respectively, over a follow-up period of 7 years.110 Malignant tachyarrhythmias have been suggested as the cause of SCD in such patients. Transient complete heart block is relatively common after both aortic (17.6%) and mitral (13%) valve replacement, pointing to bradyarrhythmias as another potential precipitating factor for SCD111 (see Chap. 76).
The association between mitral valve prolapse (MVP) and SCD is controversial. The prevalence of MVP is sufficiently high that its presence may just be a coincidental finding in victims of SCD. In a prospective 8-year study of 237 asymptomatic or minimally symptomatic patients with echocardiographically documented MVP, survival was not significantly different from that for a matched control population.112 On the other hand, MVP may not always be benign. Patients with MVP associated with mitral regurgitation and left ventricular dysfunction clearly carry a risk for such complications as infective endocarditis, cerebroembolic events, and SCD.113 In a significant number of victims of SCD, especially young women, MVP is the only structural cardiac disease identified.114
Several echocardiographic risk factors for SCD have been identified in asymptomatic or mildly symptomatic MVP patients without significant mitral regurgitation, including increased mitral valve annular circumference, thickness of the anterior and posterior mitral valve leaflets, presence and extent of endocardial plaque, and presence or absence of redundant mitral valve leaflets on M-mode echocardiography.112,115 Late gadolinium enhancement of the papillary muscles by cardiac magnetic resonance imaging in patients with MVP was found to be associated with complex ventricular arrhythmias, although the relationship between this finding and SCD has yet to be determined.116 The most important prognostic indicators for sudden death in MVP are probably a history of syncope and a family history of sudden death at a young age, as well as significant mitral regurgitation, abnormal left ventricular function, and a long QT interval115 (see Chap. 77).
Any inflammatory disease can cause SCD due to either ventricular tachyarrhythmias or complete heart block. Histologic findings suggestive of myocarditis have been reported in 10% to 44% of young victims of SCD.117 In adults, the diagnosis of myocarditis is made much less frequently, perhaps because of concurrent structural heart disease or because the late manifestations of the disease are indistinguishable from idiopathic dilated cardiomyopathy (see Chap. 35). In South America, however, myocarditis due to specific pathogens, such as Chagas disease, is the most frequent cause of cardiomyopathy and related SCD.118 Patients with infective endocarditis may also be at risk for SCD due to acute coronary emboli from valvular vegetations. More often, SCD is caused by acute hemodynamic deterioration due to valvular failure. Intramyocardial abscesses can also precipitate VT and lead to SCD.
Infiltrative cardiomyopathies, such as primary or secondary amyloidosis, hemochromatosis, or sarcoidosis, have been associated with predominantly cardiac conduction defects but also ventricular tachyarrhythmias and SCD. VT is sometimes the mode of presentation of sarcoidosis, can usually be reproduced by programmed electrical stimulation, and is associated with a high rate of recurrent arrhythmia and SCD119 (see Chap. 34).
A 25- to 100-fold increased risk of SCD due to arrhythmia, increasing primarily in the second postoperative decade, has been found predominantly in four congenital conditions: tetralogy of Fallot, transposition of the great arteries, aortic stenosis, and pulmonary vascular obstruction.120 In patients who have undergone reparative surgery for tetralogy of Fallot, a QRS duration of 180 ms or more was found to be the most sensitive predictor of SCD and ventricular tachyarrhythmias in 793 adults and correlated with other parameters of right ventricular volume overload. Older age at repair also increased the risk of arrhythmia.121 Electrophysiology testing has recently been found to be useful in evaluating the risk of ventricular arrhythmias in patients late after surgical repair of tetralogy of Fallot,122 as these patients can be uniquely susceptible to reentrant VT.
Transposition of the great arteries (post-Mustard and Senning procedures) is associated with at least 6% risk of late SCD, which is due in some cases to sinus node dysfunction and in others to ventricular tachyarrhythmias123 (see Chap. 47). Arterial-switch surgery for transposition may result in fewer late-term arrhythmias.
SCD is often (45%-60%) the mode of death in patients with primary or secondary pulmonary hypertension (see Chap. 71). Death can be precipitated by general anesthesia, dehydration, exertion, or pregnancy. Any process that decreases systemic vascular resistance increases right-to-left shunting and decreases pulmonary flow. The resultant peripheral desaturation may trigger lethal arrhythmias and SCD.124
The SCD risk in congenital aortic stenosis is estimated to be 1% and occurs predominantly in symptomatic patients with severe left ventricular hypertrophy. Ebstein anomaly is frequently (up to 25%) associated with the presence of accessory pathways and the Wolff-Parkinson-White syndrome, which carries a small risk of SCD (see below). Congenital heart block without associated structural heart disease occurs in 1 of 20,000 infants, and a moderate decrease in heart rate is usually well tolerated. A maternal risk factor is systemic lupus erythematosus. As previously noted, patients with severe bradycardia, however, have a tendency to develop ventricular arrhythmias. Pacemaker therapy has virtually eliminated the risk of SCD in this population.124
Sudden cardiac death is one of the hallmarks of the congenital long-QT syndrome (LQTS), a group of genetically distinct disorders resulting from mutations in 1 of 7 genes encoding cardiac ion channels or auxiliary ion-channel subunits125,126 (see also Chap. 36). The long QT interval reflects abnormal prolongation of repolarization caused by defects in outward currents (potassium) or impaired inactivation of inward currents (sodium). Prolonged repolarization enhances the propensity to develop early after-depolarizations leading to triggered activity that is the initiating mechanism for torsade de pointes (see Chap. 38). Other characteristics of this disorder, in addition to the prolonged QT interval (>440 ms for men or >460 ms for women, corrected for heart rate), include abnormal T wave contours, relative sinus bradycardia, a family history of early sudden death, and a propensity for recurrent syncope and sudden cardiac death due to polymorphic ventricular tachycardia (torsade de pointes) and VF. The autosomal recessive Jervell-Lange-Nielsen syndrome is a rare condition associated with SCD and congenital deafness.
More than 90% of the congenital forms of LQTS have been linked to specific chromosomal defects, resulting in a genetically based classification (LQT1 through LQT7) with important functional and prognostic implications.126 Multiple mutations have been identified in each gene, and this locus heterogeneity appears to be important prognostically. The risk of sudden death in LQTS is influenced by the duration of the QT interval, corrected for heart rate (QTc), the specific genetic defect, sex, family history, and possibly other factors.127,128
Torsade de pointes can be triggered by different stimuli, typically involving high adrenergic states such as exercise or sudden arousal (startle). Cardiac events associated with exercise, especially with swimming, dominate the clinical picture of LQTS1 patients, and auditory stimuli tend to be a trigger for arrhythmic events in LQTS2 patients.129 The frequency with which sudden deaths attributed to electrolyte or drug effects actually occur in unrecognized or subclinical carriers of LQTS genetic mutations is unknown, but these factors can precipitate torsade de pointes in known LQT patients. Registry data suggest a >50% risk by age 40 of syncope, cardiac arrest, or sudden death for LQT1, LQT2, and male LQT3 gene carriers with QTc intervals >500 ms.127 Lower levels of risk have been reported for other mutations and for patients with shorter QT intervals.
β-Blockers are a mainstay of treatment for LQTS patients, and non-randomized data suggest these drugs reduce mortality.128,130 Left-sided sympathectomy has also been used in selected cases. Implantable defibrillators are clearly indicated for sudden death survivors and may also be appropriate for LQTS patients with syncope and other high-risk features, particularly when symptoms persist despite β-blocker therapy.85
Different mutations in a few of the same genes previously shown to cause long-QT syndrome have recently been identified as causing a distinct syndrome characterized by shorter than normal corrected QT intervals (<300-320 ms) and a propensity for both atrial fibrillation and SCD at potentially very young ages.131,132 The first familial analysis identified gain-of-function mutations in the KCNH2 (or hERG) gene, which codes for the potassium channel responsible for the rapidly activating delayed rectifier (IKr) current.131 Although loss of function mutations in this gene cause LQT2, the mutations in these families were associated with short QT intervals and unusually short atrial and ventricular refractory periods at electrophysiological study.133 Since this first description, two additional genetic mutations have been reported: a de novo gain of function mutation in the KCNQ1 gene (responsible for the α subunit of the KvLQT1 protein [IKs]),134 and a mutation in the KCNJ2 gene (coding the inwardly rectifying Kir2.1 [Ik1] channel), associated with a unique T wave morphology.135 Based on these preliminary data, short-QT syndrome appears to be another uncommon genetically based condition responsible for some sudden deaths in infancy, childhood, and early adulthood.132 The frequency, full range of genotypes and phenotypes, and optimal therapy of this condition will require further study to define (see Chap. 36).
Several additional types of idiopathic polymorphic VTs have been described and are associated with an unfavorable prognosis. These arrhythmias include idiopathic VF, torsade de pointes with normal QT and a short coupling interval,136 and catecholaminergic polymorphic ventricular tachycardia. They can occur in sporadic or familial forms and are frequently though not uniformly associated with catecholamine release during physical or emotional stress.
Catecholaminergic polymorphic VT, characterized initially by a pattern of bidirectional VT first described in children, has been linked to mutations in the cardiac ryanodine receptor (RyR2)137 and calsequestrin (CASQ2)138 genes, both of which encode proteins involved in the handling of calcium by the sarcoplasmic reticulum. Patients with RyR2 mutations have also presented with polymorphic VT and VF in adulthood. These arrhythmias are at least partly responsive to β-blockers.137 The pathophysiologic mechanisms underlying short-coupled torsade de pointes and other forms of idiopathic VF have not yet been characterized.
Although the list of potential causes of SCD continues to grow, a definite cause cannot be established in a minority of patients suffering cardiac arrest. These instances of SCD without evident cause are sometimes attributed to idiopathic VF. The incidence of idiopathic VF is higher in selected populations such as younger patients (up to 14% in patients <40 years of age)139) or female survivors of SCD unrelated to myocardial infarction (10%19). The risk of recurrent VF in this young and otherwise healthy patient population ranges between 22% and 37% at 2 to 4 years.140 In survivors of cardiac arrest due to idiopathic VF, the diagnosis is made by exclusion if extensive cardiac workup reveals no diagnostic abnormality. An intriguing recent study found that an early repolarization pattern—heretofore believed to be a benign or “normal variant”—is present on ECG in an unexpectedly high number of “idiopathic VF” cases, mostly men.141 Because most people with these ECG findings do not die suddenly, the mechanistic implication of these findings is not presently clear.
In a recent analysis of 63 survivors of cardiac arrest with no evident cardiac disease, investigators followed a systematic evaluation that included advanced imaging, signal averaged ECG, exercise testing, provocative drug testing, and focused genetic testing. Using this approach, 24 patients were definitively diagnosed with primary electrical disease. The highest yield tests were pharmacologic challenge and exercise testing, particularly in establishing the diagnoses of long QT syndromes and catecholaminergic polymorphic VT. This type of thorough systematic testing may help establish diagnoses in cases of sudden death presumed to be a result of idiopathic VF.142
The syndrome of SCD associated with complete or incomplete right bundle-branch block and ST-segment elevation in V1 to V3 in patients without demonstrable structural heart disease is known as the Brugada syndrome.143
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


