ST-SEGMENT ELEVATION MYOCARDIAL INFARCTION
Elliott M. Antman ![]() Joseph Loscalzo
Joseph Loscalzo
Acute myocardial infarction (AMI) is one of the most common diagnoses in hospitalized patients in industrialized countries. In the United States, approximately 650,000 patients experience a new AMI and 450,000 experience a recurrent AMI each year. The early (30-day) mortality rate from AMI is ~30%, with more than half of these deaths occurring before the stricken individual reaches the hospital. Although the mortality rate after admission for AMI has declined by ~30% over the past two decades, approximately 1 of every 25 patients who survives the initial hospitalization dies in the first year after AMI. Mortality is approximately fourfold higher in elderly patients (over age 75) as compared with younger patients.
When patients with prolonged ischemic discomfort at rest are first seen, the working clinical diagnosis is that they are suffering from an acute coronary syndrome (Fig. 35-1). The 12-lead electrocardiogram (ECG) is a pivotal diagnostic and triage tool because it is at the center of the decision pathway for management; it permits distinction of those patients presenting with ST-segment elevation from those presenting without ST-segment elevation. Serum cardiac biomarkers are obtained to distinguish unstable angina (UA) from non-ST-segment MI (NSTEMI) and to assess the magnitude of an ST-segment elevation MI (STEMI). This chapter focuses on the evaluation and management of patients with STEMI, while Chap. 34 discusses UA/NSTEMI.
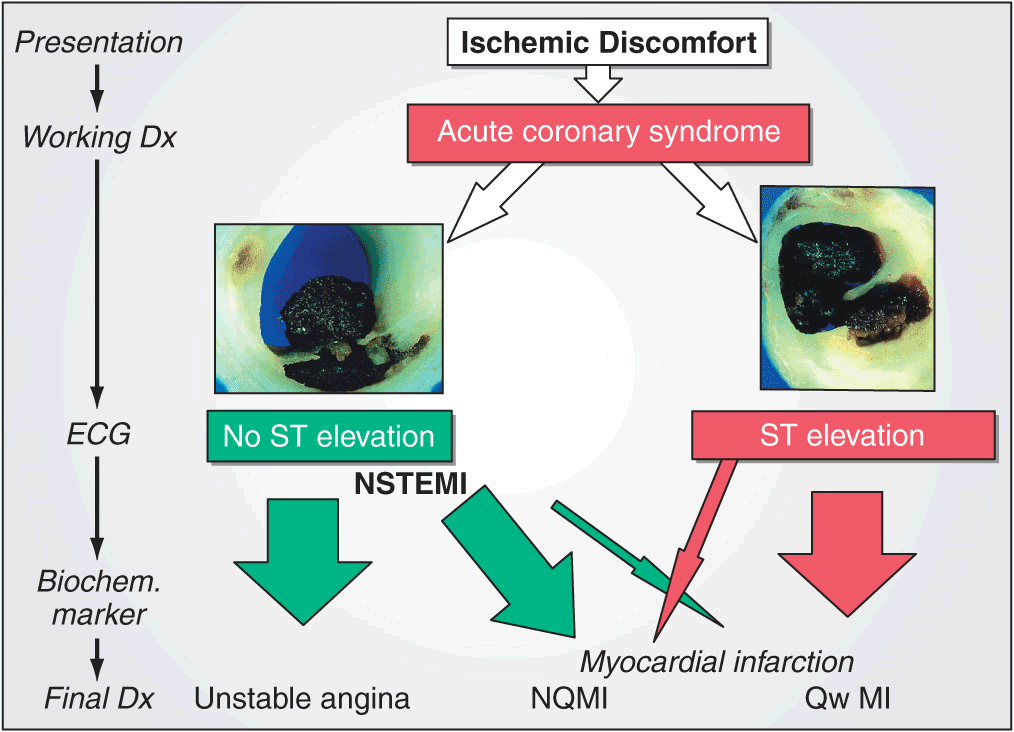
FIGURE 35-1
Acute coronary syndromes. Following disruption of a vulnerable plaque, patients experience ischemic discomfort resulting from a reduction of flow through the affected epicardial coronary artery. The flow reduction may be caused by a completely occlusive thrombus (right) or subtotally occlusive thrombus (left). Patients with ischemic discomfort may present with or without ST-segment elevation. Of patients with ST-segment elevation, the majority (wide red arrow) ultimately develop a Q wave on the ECG (QwMI), while a minority (thin red arrow) do not develop Q wave and, in older literature, were said to have sustained a non-Q-wave MI (NQMI). Patients who present without ST-segment elevation are suffering from either unstable angina or a non-ST-segment elevation MI (NSTEMI) (wide green arrows), a distinction that is ultimately made on the presence or absence of a serum cardiac marker such as CKMB or a cardiac troponin detected in the blood. The majority of patients presenting with NSTEMI do not develop a Q wave on the ECG; a minority develop a QwMI (thin green arrow). (Adapted from CW Hamm et al: Lancet 358:1533, 2001, and MJ Davies: Heart 83:361, 2000; with permission from the BMJ Publishing Group.)
PATHOPHYSIOLOGY: ROLE OF ACUTE PLAQUE RUPTURE
STEMI usually occurs when coronary blood flow decreases abruptly after a thrombotic occlusion of a coronary artery previously affected by atherosclerosis.
Slowly developing, high-grade coronary artery stenoses do not typically precipitate STEMI because of the development of a rich collateral network over time. Instead, STEMI occurs when a coronary artery thrombus develops rapidly at a site of vascular injury. This injury is produced or facilitated by factors such as cigarette smoking, hypertension, and lipid accumulation. In most cases, STEMI occurs when the surface of an atherosclerotic plaque becomes disrupted (exposing its contents to the blood) and conditions (local or systemic) favor thrombogenesis. A mural thrombus forms at the site of plaque disruption, and the involved coronary artery becomes occluded. Histologic studies indicate that the coronary plaques prone to disruption are those with a rich lipid core and a thin fibrous cap (Chap. 30). After an initial platelet monolayer forms at the site of the disrupted plaque, various agonists (collagen, ADP, epinephrine, serotonin) promote platelet activation. After agonist stimulation of platelets, thromboxane A2 (a potent local vasoconstrictor) is released, further platelet activation occurs, and potential resistance to fibrinolysis develops.
In addition to the generation of thromboxane A2, activation of platelets by agonists promotes a conformational change in the glycoprotein IIb/IIIa receptor. Once converted to its functional state, this receptor develops a high affinity for soluble adhesive proteins (i.e., integrins) such as fibrinogen. Since fibrinogen is a multivalent molecule, it can bind to two different platelets simultaneously, resulting in platelet cross-linking and aggregation.
The coagulation cascade is activated on exposure of tissue factor in damaged endothelial cells at the site of the disrupted plaque. Factors VII and X are activated, ultimately leading to the conversion of prothrombin to thrombin, which then converts fibrinogen to fibrin. Fluid-phase and clot-bound thrombin participate in an autoamplification reaction leading to further activation of the coagulation cascade. The culprit coronary artery eventually becomes occluded by a thrombus containing platelet aggregates and fibrin strands.
In rare cases, STEMI may be due to coronary artery occlusion caused by coronary emboli, congenital abnormalities, coronary spasm, and a wide variety of systemic—particularly inflammatory—diseases. The amount of myocardial damage caused by coronary occlusion depends on (1) the territory supplied by the affected vessel, (2) whether or not the vessel becomes totally occluded, (3) the duration of coronary occlusion, (4) the quantity of blood supplied by collateral vessels to the affected tissue, (5) the demand for oxygen of the myocardium whose blood supply has been suddenly limited, (6) endogenous factors that can produce early spontaneous lysis of the occlusive thrombus, and (7) the adequacy of myocardial perfusion in the infarct zone when flow is restored in the occluded epicardial coronary artery.
Patients at increased risk for developing STEMI include those with multiple coronary risk factors (Chap. 30) and those with unstable angina (Chap. 34). Less common underlying medical conditions predisposing patients to STEMI include hypercoagulability, collagen vascular disease, cocaine abuse, and intracardiac thrombi or masses that can produce coronary emboli.
There have been major advances in the management of STEMI with recognition that the “chain of survival” involves a highly integrated system starting with prehospital care and extending to early hospital management so as to provide expeditious implementation of a reperfusion strategy.
CLINICAL PRESENTATION
In up to one-half of cases, a precipitating factor appears to be present before STEMI, such as vigorous physical exercise, emotional stress, or a medical or surgical illness. Although STEMI may commence at any time of the day or night, circadian variations have been reported such that clusters are seen in the morning within a few hours of awakening.
Pain is the most common presenting complaint in patients with STEMI. The pain is deep and visceral; adjectives commonly used to describe it are heavy, squeezing, and crushing, although, occasionally, it is described as stabbing or burning (Chap. 4). It is similar in character to the discomfort of angina pectoris (Chap. 33) but commonly occurs at rest, is usually more severe, and lasts longer. Typically, the pain involves the central portion of the chest and/or the epigastrium, and, on occasion, it radiates to the arms. Less common sites of radiation include the abdomen, back, lower jaw, and neck. The frequent location of the pain beneath the xiphoid and epigastrium and the patients’ denial that they may be suffering a heart attack are chiefly responsible for the common mistaken impression of indigestion. The pain of STEMI may radiate as high as the occipital area but not below the umbilicus. It is often accompanied by weakness, sweating, nausea, vomiting, anxiety, and a sense of impending doom. The pain may commence when the patient is at rest, but when it begins during a period of exertion, it does not usually subside with cessation of activity, in contrast to angina pectoris.
The pain of STEMI can simulate pain from acute pericarditis (Chap. 22), pulmonary embolism, acute aortic dissection (Chap. 38), costochondritis, and gastrointestinal disorders. These conditions should therefore be considered in the differential diagnosis. Radiation of discomfort to the trapezius is not seen in patients with STEMI and may be a useful distinguishing feature that suggests pericarditis is the correct diagnosis. However, pain is not uniformly present in patients with STEMI. The proportion of painless STEMIs is greater in patients with diabetes mellitus, and it increases with age. In the elderly, STEMI may present as sudden-onset breathlessness, which may progress to pulmonary edema. Other less common presentations, with or without pain, include sudden loss of consciousness, a confusional state, a sensation of profound weakness, the appearance of an arrhythmia, evidence of peripheral embolism, or merely an unexplained drop in arterial pressure.
PHYSICAL FINDINGS
Most patients are anxious and restless, attempting unsuccessfully to relieve the pain by moving about in bed, altering their position, and stretching. Pallor associated with perspiration and coolness of the extremities occurs commonly. The combination of substernal chest pain persisting for >30 min and diaphoresis strongly suggests STEMI. Although many patients have a normal pulse rate and blood pressure within the first hour of STEMI, about one-fourth of patients with anterior infarction have manifestations of sympathetic nervous system hyperactivity (tachycardia and/or hypertension), and up to one-half with inferior infarction show evidence of parasympathetic hyperactivity (bradycardia and/or hypotension).
The precordium is usually quiet, and the apical impulse may be difficult to palpate. In patients with anterior wall infarction, an abnormal systolic pulsation caused by dyskinetic bulging of infarcted myocardium may develop in the periapical area within the first days of the illness and then may resolve. Other physical signs of ventricular dysfunction include fourth and third heart sounds, decreased intensity of the first heart sound, and paradoxical splitting of the second heart sound (Chap. 9). A transient mid-systolic or late systolic apical systolic murmur due to dysfunction of the mitral valve apparatus may be present. A pericardial friction rub is heard in many patients with transmural STEMI at some time in the course of the disease, if the patients are examined frequently. The carotid pulse is often decreased in volume, reflecting reduced stroke volume. Temperature elevations up to 38°C may be observed during the first week after STEMI. The arterial pressure is variable; in most patients with transmural infarction, systolic pressure declines by approximately 10–15 mmHg from the preinfarction state.
LABORATORY FINDINGS
Myocardial infarction (MI) progresses through the following temporal stages: (1) acute (first few hours– 7 days), (2) healing (7–28 days), and (3) healed (29 days). When evaluating the results of diagnostic tests for STEMI, the temporal phase of the infarction must be considered. The laboratory tests of value in confirming the diagnosis may be divided into four groups: (1) ECG, (2) serum cardiac biomarkers, (3) cardiac imaging, and (4) nonspecific indices of tissue necrosis and inflammation.
ELECTROCARDIOGRAM
The electrocardiographic manifestations of STEMI are described in Chap. 11. During the initial stage, total occlusion of an epicardial coronary artery produces ST-segment elevation. Most patients initially presenting with ST-segment elevation ultimately evolve Q waves on the ECG. However, Q waves in the leads overlying the infarct zone may vary in magnitude and even appear only transiently, depending on the reperfusion status of the ischemic myocardium and restoration of trans-membrane potentials over time. A small proportion of patients initially presenting with ST-segment elevation will not develop Q waves when the obstructing thrombus is not totally occlusive, obstruction is transient, or if a rich collateral network is present. Among patients presenting with ischemic discomfort but without ST-segment elevation, if a serum cardiac biomarker of necrosis (see later) is detected, the diagnosis of NSTEMI is ultimately made (Fig. 35-1). A minority of patients who present initially without ST-segment elevation may develop a Q-wave MI. Previously, it was believed that transmural MI is present if the ECG demonstrates Q waves or loss of R waves, and nontransmural MI may be present if the ECG shows only transient ST-segment and T-wave changes. However, electrocardiographicpathologic correlations are far from perfect and terms such as Q-wave MI, non-Q-wave MI, transmural MI, and nontransmural MI, have been replaced by STEMI and NSTEMI (Fig. 35-1). Contemporary studies using MRI suggest that the development of a Q wave on the ECG is more dependent on the volume of infarcted tissue rather than the transmurality of infarction.
SERUM CARDIAC BIOMARKERS
Certain proteins, called serum cardiac biomarkers, are released from necrotic heart muscle after STEMI. The rate of liberation of specific proteins differs depending on their intracellular location, their molecular weight, and the local blood and lymphatic flow. Cardiac biomarkers become detectable in the peripheral blood once the capacity of the cardiac lymphatics to clear the interstitium of the infarct zone is exceeded and spillover into the venous circulation occurs. The temporal pattern of protein release is of diagnostic importance, but contemporary urgent reperfusion strategies necessitate making a decision (based largely on a combination of clinical and ECG findings) before the results of blood tests have returned from the laboratory. Rapid whole-blood bedside assays for serum cardiac markers are now available and may facilitate management decisions, particularly in patients with nondiagnostic ECGs.
Cardiac-specific troponin T (cTnT) and cardiac-specific troponin I (cTnI) have amino-acid sequences different from those of the skeletal muscle forms of these proteins. These differences permitted the development of quantitative assays for cTnT and cTnI with highly specific monoclonal antibodies. Since cTnT and cTnI are not normally detectable in the blood of healthy individuals but may increase after STEMI to levels >20 times higher than the upper reference limit (the highest value seen in 99% of a reference population not suffering from MI), the measurement of cTnT or cTnI is of considerable diagnostic usefulness, and they are now the preferred biochemical markers for MI (Fig. 35-2). The cardiac troponins are particularly valuable when there is clinical suspicion of either skeletal muscle injury or a small MI that may be below the detection limit for creatine phosphokinase (CK) and its MB isoenzyme (CKMB) measurements, and they are, therefore, of particular value in distinguishing UA from NSTEMI. Levels of cTnI and cTnT may remain elevated for 7–10 days after STEMI.
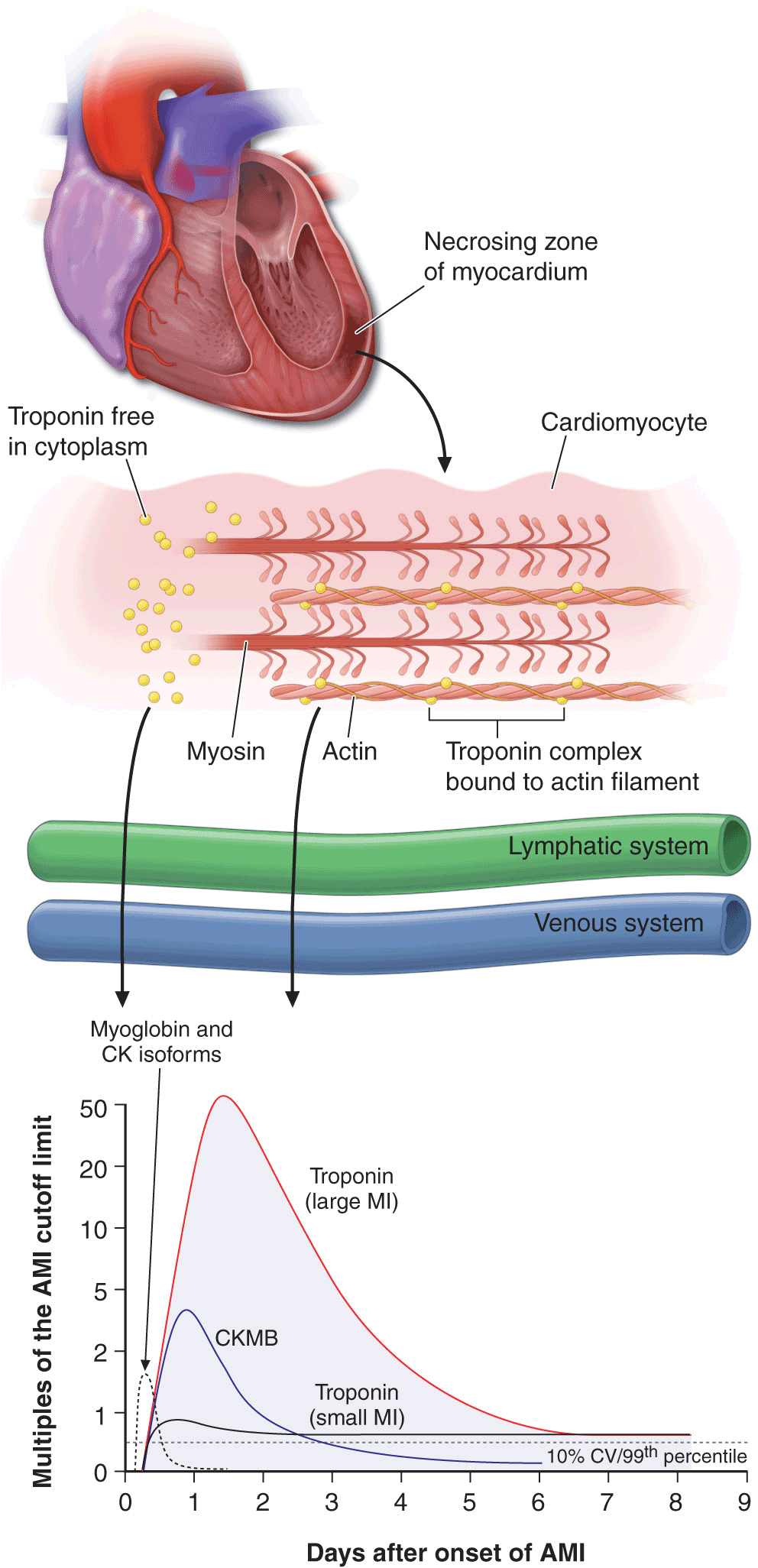
FIGURE 35-2
The zone of necrosing myocardium is shown at the top of the figure, followed in the middle portion of the figure by a diagram of a cardiomyocyte that is in the process of releasing biomarkers. The biomarkers that are released into the interstitium are first cleared by lymphatics followed subsequently by spillover into the venous system. After disruption of the sarcolemmal membrane of the cardiomyocyte, the cytoplasmic pool of biomarkers is released first (left-most arrow in bottom portion of figure). Markers such as myoglobin and CK isoforms are rapidly released, and blood levels rise quickly above the cutoff limit; this is then followed by a more protracted release of biomarkers from the disintegrating myofilaments that may continue for several days. Cardiac troponin levels rise to about 20 to 50 times the upper reference limit (the 99th percentile of values in a reference control group) in patients who have a “classic” acute myocardial infarction (MI) and sustain sufficient myocardial necrosis to result in abnormally elevated levels of the MB fraction of creatine kinase (CKMB). Clinicians can now diagnose episodes of microinfarction by sensitive assays that detect cardiac troponin elevations above the upper reference limit, even though CKMB levels may still be in the normal reference range (not shown). CV = coefficient of variation. (Modified from EM Antman: Decision making with cardiac troponin tests. N Engl J Med 346:2079, 2002 and AS Jaffe et al: Biomarkers in acute cardiac disease: The present and the future. J Am Coll Cardiol 48:1, 2006.)
CK rises within 4–8 h and generally returns to normal by 48–72 h (Fig. 35-2). An important drawback of total CK measurement is its lack of specificity for STEMI, as CK may be elevated with skeletal muscle disease or trauma, including intramuscular injection.
The MB isoenzyme of CK has the advantage over total CK that it is not present in significant concentrations in extracardiac tissue and, therefore, is considerably more specific. However, cardiac surgery, myocarditis, and electrical cardioversion often result in elevated serum levels of the MB isoenzyme. A ratio (relative index) of CKMB mass: CK activity ≥2.5 suggests but is not diagnostic of a myocardial rather than a skeletal muscle source for the CKMB elevation.
Many hospitals are using cTnT or cTnI rather than CKMB as the routine serum cardiac marker for diagnosis of STEMI, although any of these analytes remain clinically acceptable. It is not cost-effective to measure both a cardiac-specific troponin and CKMB at all time points in every patient.
While it has long been recognized that the total quantity of protein released correlates with the size of the infarct, the peak protein concentration correlates only weakly with infarct size. Recanalization of a coronary artery occlusion (either spontaneously or by mechanical or pharmacologic means) in the early hours of STEMI causes earlier peaking of biomarker measurements (Fig. 35-2) because of a rapid washout from the interstitium of the infarct zone, quickly overwhelming lymphatic clearance of the proteins.
The nonspecific reaction to myocardial injury is associated with polymorphonuclear leukocytosis, which appears within a few hours after the onset of pain and persists for 3–7 days; the white blood cell count often reaches levels of 12,000–15,000/μL. The erythrocyte sedimentation rate rises more slowly than the white blood cell count, peaking during the first week and sometimes remaining elevated for 1 or 2 weeks.
CARDIAC IMAGING
Abnormalities of wall motion on two-dimensional echo-cardiography (Chap. 12) are almost universally present. Although acute STEMI cannot be distinguished from an old myocardial scar or from acute severe ischemia by echocardiography, the ease and safety of the procedure make its use appealing as a screening tool in the emergency department setting. When the ECG is not diagnostic of STEMI, early detection of the presence or absence of wall motion abnormalities by echocardiography can aid in management decisions, such as whether the patient should receive reperfusion therapy (e.g., fibrinolysis or a percutaneous coronary intervention [PCI]). Echocardiographic estimation of left ventricular (LV) function is useful prognostically; detection of reduced function serves as an indication for therapy with an inhibitor of the renin-angiotensin-aldosterone system. Echocardiography may also identify the presence of right ventricular (RV) infarction, ventricular aneurysm, pericardial effusion, and LV thrombus. In addition, Doppler echocardiography is useful in the detection and quantitation of a ventricular septal defect and mitral regurgitation, two serious complications of STEMI.
Several radionuclide imaging techniques (Chap. 12) are available for evaluating patients with suspected STEMI. However, these imaging modalities are used less often than echocardiography because they are more cumbersome and lack sensitivity and specificity in many clinical circumstances. Myocardial perfusion imaging with [201Tl] or [99mTc]-sestamibi, which are distributed in proportion to myocardial blood flow and concentrated by viable myocardium (Chap. 33), reveal a defect (“cold spot”) in most patients during the first few hours after development of a transmural infarct. Although perfusion scanning is extremely sensitive, it cannot distinguish acute infarcts from chronic scars and, thus, is not specific for the diagnosis of acute MI. Radionuclide ventriculography, carried out with [99mTc]-labeled red blood cells, frequently demonstrates wall motion disorders and reduction in the ventricular ejection fraction in patients with STEMI. While of value in assessing the hemodynamic consequences of infarction and in aiding in the diagnosis of RV infarction when the RV ejection fraction is depressed, this technique is nonspecific, as many cardiac abnormalities other than MI alter the radionuclide ventriculogram.
Myocardial infarction can be detected accurately with high-resolution cardiac MRI (Chap. 12) using a technique referred to as late enhancement. A standard imaging agent (gadolinium) is administered and images are obtained after a 10-min delay. Since little gadolinium enters normal myocardium, where there are tightly packed myocytes, but does percolate into the expanded intercellular region of the infarct zone, there is a bright signal in areas of infarction that appears in stark contrast to the dark areas of normal myocardium.
INITIAL MANAGEMENT
PREHOSPITAL CARE
The prognosis in STEMI is largely related to the occurrence of two general classes of complications: (1) electrical complications (arrhythmias) and (2) mechanical complications (“pump failure”). Most out-of-hospital deaths from STEMI are due to the sudden development of ventricular fibrillation. The vast majority of deaths due to ventricular fibrillation occur within the first 24 h of the onset of symptoms, and of these, over half occur in the first hour. Therefore, the major elements of pre-hospital care of patients with suspected STEMI include (1) recognition of symptoms by the patient and prompt seeking of medical attention; (2) rapid deployment of an emergency medical team capable of performing resuscitative maneuvers, including defibrillation; (3) expeditious transportation of the patient to a hospital facility that is continuously staffed by physicians and nurses skilled in managing arrhythmias and providing advanced cardiac life support; and (4) expeditious implementation of reperfusion therapy (Fig. 35-3). The greatest delay usually occurs not during transportation to the hospital but, rather, between the onset of pain and the patient’s decision to call for help. This delay can best be reduced by health care professionals educating the public concerning the significance of chest discomfort and the importance of seeking early medical attention. Regular office visits with patients having a history of or who are at risk for ischemic heart disease are important “teachable moments” for clinicians to review the symptoms of STEMI and the appropriate action plan.
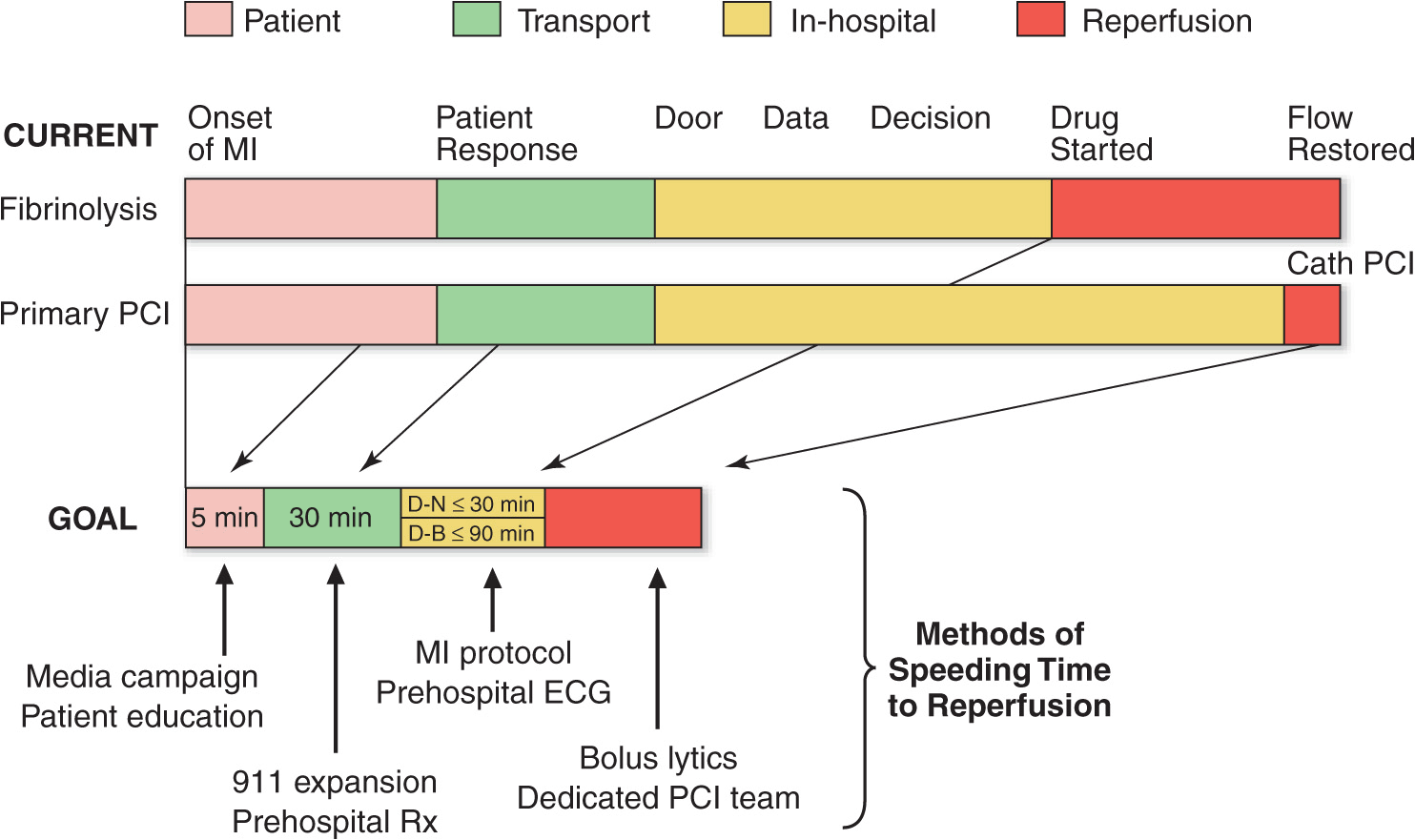
FIGURE 35-3
Major components of time delay between onset of symptoms from STEMI and restoration of flow in the infarct-related artery. Plotted sequentially from left to right are the times for patients to recognize symptoms and seek medical attention, transportation to the hospital, in-hospital decision making, implementation of reperfusion strategy, and restoration of flow once the reperfusion strategy has been initiated. The time to initiate fibrinolytic therapy is the “door-to-needle” (D-N) time; this is followed by the period of time required for pharmacologic restoration of flow. More time is required to move the patient to the catheterization laboratory for a percutaneous coronary interventional (PCI) procedure, referred to as the “door-to-balloon” (D-B) time, but restoration of flow in the epicardial infarct–related artery occurs promptly after PCI. At the bottom is a variety of methods for speeding the time to reperfusion along with the goals for the time intervals for the various components of the time delay. (Adapted from CP Cannon et al: J Thromb Thrombol 1:27, 1994.)
Increasingly, monitoring and treatment are carried out by trained personnel in the ambulance, further shortening the time between the onset of the infarction and appropriate treatment. General guidelines for initiation of fibrinolysis in the prehospital setting include the ability to transmit 12-lead ECGs to confirm the diagnosis, the presence of paramedics in the ambulance, training of paramedics in the interpretation of ECGs and management of STEMI, and online medical command and control that can authorize the initiation of treatment in the field.
MANAGEMENT IN THE EMERGENCY DEPARTMENT
In the emergency department, the goals for the management of patients with suspected STEMI include control of cardiac discomfort, rapid identification of patients who are candidates for urgent reperfusion therapy, triage of lower-risk patients to the appropriate location in the hospital, and avoidance of inappropriate discharge of patients with STEMI. Many aspects of the treatment of STEMI are initiated in the emergency department and then continued during the in-hospital phase of management.
Aspirin is essential in the management of patients with suspected STEMI and is effective across the entire spectrum of acute coronary syndromes (Fig. 35-1). Rapid inhibition of cyclooxygenase-1 in platelets followed by a reduction of thromboxane A2 levels is achieved by buccal absorption of a chewed 160–325-mg tablet in the emergency department. This measure should be followed by daily oral administration of aspirin in a dose of 75–162 mg.
In patients whose arterial O2 saturation is normal, supplemental O2 is of limited if any clinical benefit and therefore is not cost-effective. However, when hypoxemia is present, O2 should be administered by nasal prongs or face mask (2–4 L/min) for the first 6–12 h after infarction; the patient should then be reassessed to determine if there is a continued need for such treatment.
CONTROL OF DISCOMFORT
Sublingual nitroglycerin can be given safely to most patients with STEMI. Up to three doses of 0.4 mg should be administered at about 5-min intervals. In addition to diminishing or abolishing chest discomfort, nitroglycerin may be capable of both decreasing myocardial oxygen demand (by lowering preload) and increasing myocardial oxygen supply (by dilating infarct-related coronary vessels or collateral vessels). In patients whose initially favorable response to sublingual nitroglycerin is followed by the return of chest discomfort, particularly if accompanied by other evidence of ongoing ischemia such as further ST-segment or T-wave shifts, the use of intravenous nitroglycerin should be considered. Therapy with nitrates should be avoided in patients who present with low systolic arterial pressure (<90 mmHg) or in whom there is clinical suspicion of right ventricular infarction (inferior infarction on ECG, elevated jugular venous pressure, clear lungs, and hypotension). Nitrates should not be administered to patients who have taken the phosphodiesterase-5 inhibitor sildenafil for erectile dysfunction within the preceding 24 h, because it may potentiate the hypotensive effects of nitrates. An idiosyncratic reaction to nitrates, consisting of sudden marked hypotension, sometimes occurs but can usually be reversed promptly by the rapid administration of intravenous atropine.
Morphine is a very effective analgesic for the pain associated with STEMI. However, it may reduce sympathetically mediated arteriolar and venous constriction, and the resulting venous pooling may reduce cardiac output and arterial pressure. These hemodynamic disturbances usually respond promptly to elevation of the legs, but in some patients volume expansion with intravenous saline is required. The patient may experience diaphoresis and nausea, but these events usually pass and are replaced by a feeling of well-being associated with the relief of pain. Morphine also has a vagotonic effect and may cause bradycardia or advanced degrees of heart block, particularly in patients with inferior infarction. These side effects usually respond to atropine (0.5 mg intravenously). Morphine is routinely administered by repetitive (every 5 min) intravenous injection of small doses (2–4 mg), rather than by the subcutaneous administration of a larger quantity, because absorption may be unpredictable by the latter route.
Intravenous beta blockers are also useful in the control of the pain of STEMI. These drugs control pain effectively in some patients, presumably by diminishing myocardial O2 demand and hence ischemia. More important, there is evidence that intravenous beta blockers reduce the risks of reinfarction and ventricular fibrillation (see “Beta-Adrenoceptor Blockers,” later). However, patient selection is important when considering beta blockers for STEMI. Oral Betablocker therapy should be initiated in the first 24 h for patients who do not have any of the following: (1) signs of heart failure, (2) evidence of a low-output state, (3) increased risk for cardiogenic shock, or (4) other relative contraindications to beta blockade (PR interval greater than 0.24 s, second- or third-degree heart block, active asthma, or reactive airway disease). A commonly employed regimen is metoprolol, 5 mg every 2–5 min for a total of 3 doses, provided the patient has a heart rate >60 beats per minute (bpm), systolic pressure >100 mmHg, a PR interval <0.24 s, and rales that are no higher than 10 cm up from the diaphragm. Fifteen min after the last intravenous dose, an oral regimen is initiated of 50 mg every 6 h for 48 h, followed by 100 mg every 12 h.
Unlike beta blockers, calcium antagonists are of little value in the acute setting, and there is evidence that short-acting dihydropyridines may be associated with an increased mortality risk.
MANAGEMENT STRATEGIES
The primary tool for screening patients and making triage decisions is the initial 12-lead ECG. When ST-segment elevation of at least 2 mm in two contiguous precordial leads and 1 mm in two adjacent limb leads is present, a patient should be considered a candidate for reperfusion therapy (Fig. 35-4). The process of selecting patients for fibrinolysis versus primary PCI (angioplasty, or stenting; Chap. 36) is discussed later. In the absence of ST-segment elevation, fibrinolysis is not helpful, and evidence exists suggesting that it may be harmful.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


