Rheumatologic Diseases and the Cardiovascular System: Introduction
Rheumatologic conditions such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and the vasculitides that affect multiple organ systems may also impact the cardiovascular system. These pathologic processes fall in the category of autoimmune diseases, which are initiated by a complex interplay between genetic factors and environmental stimuli. They are presumed to be driven by self-reactive T and B lymphocytes, which in tandem with a network of endogenous and exogenous signals, activate the immune system, producing tissue inflammation and damage (Table 89–1). The effects of autoimmunity on the cardiovascular system may be the result of local or systemic mechanisms. For example, locally aberrant immunity may selectively target the pericardium, myocardium, or conduction system in systemic sclerosis. On the other hand, in patients with SLE or systemic vasculitis, circulating immune complexes may deposit in blood vessels, where they evoke an inflammatory response, which in turn occludes the vessel lumen and causes ischemic manifestations distal to the site of critically limited blood flow. A hypercoagulable state from a condition known as antiphospholipid antibody syndrome can lead to thrombotic occlusion, producing myocardial infarction, stroke, or ischemic damage of the visceral organs. Thus diverse pathways of a dysregulated immune system may converge to directly or indirectly damage the heart and vasculature.
| Disease | Sex Distribution | Clinical Manifestations | Cardiovascular Manifestations |
|---|---|---|---|
| Rheumatoid arthritis | F>M | Inflammatory polyarthritis, rheumatoid nodules, RF, anti-CCP | Pericarditis, coronary artery disease, cardiomyopathy, congestive heart failure |
| Systemic lupus erythematosus | F>M | Malar rash, arthritis, photosensitivity, serositis, nephritis, +ANA; may have concomitant APS | Pericarditis, Libman-Sacks endocarditis, coronary artery disease, hypertension |
| Inflammatory myopathies | F>M | Proximal muscle weakness, DM with Gottron papules, shawl sign, mechanics hands | Pericarditis, conduction system abnormalities, congestive heart failure, and myocarditis |
| Systemic sclerosis | F>M | Limited form is referred to as CREST. Diffuse form involves proximal skin and visceral organs | Pulmonary hypertension, pericarditis, cardiomyopathy, conduction system disease |
| Seronegative spondyloarthropathy | M>F | Spinal or sacroiliac involvement, enthesitis, absence of rheumatoid factor, and a high incidence of HLA-B27 | Aortitis, conduction system disease |
This chapter aims to familiarize the cardiovascular specialist with the clinical features of rheumatologic diseases that affect the heart and blood vessels. These diseases derive from chronic inflammation and abnormal tissue repair. This chapter also covers the most common heritable diseases of connective tissue (eg, Marfan syndrome, Ehlers-Danlos pseudoxanthoma elasticum). Unlike autoimmune diseases, these rare disorders mostly result from mutations in specific genes encoding various components of connective tissue that maintain the structural integrity of the vasculature.
Cardiovascular Manifestations of Systemic Rheumatic Diseases
With a worldwide prevalence of 0.5% to 1%,1 RA ranks as the most common of the systemic autoimmune diseases. RA is a female-predominant disease characterized by a chronic symmetrical polyarthritis with a strong predilection for the small joints of the hands and feet that often leads to physical impairment and disability. The diagnosis is established on clinical grounds by recognizing the signs and symptoms of joint inflammation. It is supported by the results of laboratory studies and plain radiographs of the hands and feet. Approximately three-quarters of patients with RA test positive for serum rheumatoid factor or anticyclic citrullinated peptide antibodies.2 X-rays of the hands and feet are often normal at the onset of RA, but later they may show the characteristic marginal joint erosions associated with this disease, as well as joint space narrowing from thinning of the articular cartilage. Much is known about the pathogenesis of synovial inflammation in RA. Briefly, the synovial tissue is characterized by a chronic inflammatory infiltrate comprised chiefly of T cells, B cells, macrophages, fibroblasts, and mast cells. There is increased production of a diverse array of proinflammatory mediators, such as tumor necrosis factor (TNF) α, interleukin (IL) 1, and IL-6, as well as many chemokines that work together to amplify the pathologic response, stimulate proliferation of synovial fibroblasts, up-regulate expression of adhesion molecules on blood vessels, and promote angiogenesis.
Although RA is predominately an inflammatory joint disease, it produces systemic effects that can target other organs and tissues. Extraarticular manifestations include fatigue, low-grade fever, Sjögren syndrome, nodules, interstitial lung disease, and vasculitis. In RA, the most common cardiovascular manifestations are pericarditis, valvular disease, cardiomyopathy, coronary vasculitis, ischemic heart disease, and congestive heart failure.
In a necropsy series, evidence of pericarditis can be seen in as many as 54% of cases.3 Acute, symptomatic pericarditis, however, occurs in fewer than 10% of patients with severe RA.4,5 RA patients with acute pericarditis are clinically indistinguishable from those with pericardial disease secondary to nonrheumatic conditions. Symptoms of positional chest pain or a pericardial friction rub may be elicited in up to 55% to 65% of patients.6,7 Although in this setting electrocardiograms are often normal, 5% to 10% of patients may manifest abnormalities classically associated with acute pericarditis. Imaging may be useful in confirming the diagnosis of pericarditis. In one series, echocardiography revealed a pericardial effusion in 90% of patients with this clinical diagnosis.6 Aspiration of the pericardial fluid, which is not usually required for diagnosis in the appropriate clinical setting, reveals an elevated white blood cell count, protein, and lactate dehydrogenase; decreased glucose levels; the presence of rheumatoid factor, and a low complement7; these features may help to differentiate it from other causes of pericarditis.
In one cohort, the occurrence of pericarditis was associated with decreased survival at 10 years.6 However, it is unlikely that the presence of pericarditis directly contributes to the increased risk of death as the presence of extraarticular disease in general is associated with increased mortality. Constrictive pericarditis, albeit rare, is associated with higher mortality rates than uncomplicated pericarditis. It requires an urgent intervention, such as pericardial window or stripping procedure.8 Constrictive pericarditis must be distinguished from restrictive cardiomyopathy, which may result from secondary amyloidosis rarely seen in patients with long-standing RA. Most patients with uncomplicated acute pericarditis will respond to treatment with nonsteroidal anti-inflammatory drugs or corticosteroids. Corticosteroid therapy is generally reserved for patients with moderate to severe pericarditis and usually consists of a short burst of high doses of prednisone (eg, 40-60 mg/d) with subsequent taper over a period of several weeks, depending on the clinical response.
Clinically significant valvular disease is relatively uncommon in RA. In autopsy reviews, only 3% to 5% of patients with RA had valvular involvement in the form of rheumatoid nodules.9,10 Patients with subcutaneous rheumatoid nodules have a higher rate of mitral valve insufficiency than those without nodules,11 but unlike patients with rheumatic heart disease, mitral valve disease associated with RA does not lead to valvular stenosis. Patients with severe or symptomatic valvular disease require surgical intervention.12,13
RA may cause a cardiomyopathy characterized by focal granulomatous inflammation of the myocardium.4 The granulomas may involve the conduction system, leading to first-, second-, or third-degree heart block, which usually persists despite immunosuppressive therapy.14 Rarely, cardiomyopathy may result from secondary amyloidosis, which manifests on echocardiograms as a significant increase in ventricular wall thickness and wall motion abnormalities.15 Cardiac MRI (cMRI) may be used to differentiate the different causes of cardiomyopathy. In particular, images from cMRI showing delayed enhancement with gadolinium display a characteristic diffuse endocardial hyperenhancement pattern that may suggest cardiac amyloidosis.16
Although as many as 20% of patients with RA show histologic evidence of coronary vasculitis by autopsy, the significance of this finding is poorly understood because it rarely occurs as a clinical complication.9 The diagnosis of coronary vasculitis involving the epicardial vessels may be suggested by the angiographic findings of interspersed areas of smooth-walled stenosis and ectasia as well as focal aneurysms. However, these angiographic features are relatively nonspecific and may be due to atherosclerosis. Recognition of coronary vasculitis is important because it requires treatment with immunosuppressive therapy such as high-dose corticosteroids and possibly other agents.17
Overall mortality is increased in patients with RA compared with the general population.18,19 It is striking that 40% of deaths in RA are attributable to cardiovascular disease.20,21 In a recent study, the prevalence of cardiovascular disease in RA patients was comparable to that of patients with diabetes.22 It is known that a significant proportion of patients with RA judged to be in clinical remission using standardized disease activity scores still show an elevated level of C-reactive protein (>3 mg/L), a known risk factor for future cardiovascular mortality.23 Increasing evidence supports a strong link between RA and accelerated atherosclerosis, highlighting it as an important risk factor for cardiovascular disease.
Atherosclerosis is an inflammatory process driven by many of the same mediators that are associated with rheumatoid inflammation.24 Systemic inflammation in RA is hypothesized to accelerate atherosclerosis, as well as affect other tissues, such as liver, muscle, and fat, which influence other cardiovascular risk factors (Fig. 89–1). Additionally, RA appears to be an independent risk factor for multivessel coronary artery disease,25 and as shown in the Nurses Health Study, women with RA have a two-fold higher rate of myocardial infarctions compared with controls.26 Other studies suggest that RA patients are less likely to be symptomatic from ischemic heart disease as non-RA controls and twice as likely to have sudden death and unrecognized myocardial infarction,27 contributing to a higher incidence of death from coronary atherosclerosis.28
Figure 89–1.
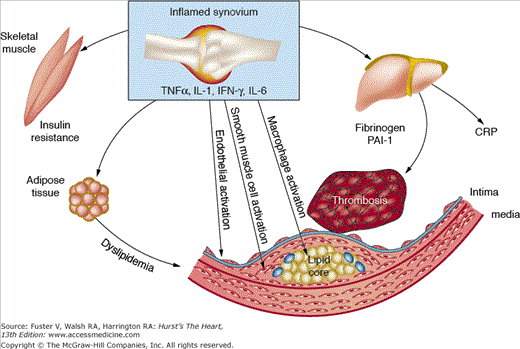
Systemic effects of inflammation in rheumatoid arthritis. The rheumatoid joint expresses high levels of various proinflammatory mediators, including tumor necrosis factor (TNF) α, interleukin (IL) 1, and IL-6, that amplify the inflammatory response. T helper cells secrete interferon-γ (IFN-γ) and IL-17, which in turn activate the cellular constituents of the synovial tissue. These cytokines, which are also found in the vascular endothelium of the atherosclerotic blood vessel, serve to promote coronary artery disease and plaque rupture. Additionally, up-regulation of these cytokines influence other cardiovascular risk factors by affecting skeletal muscle, adipose tissue, and the liver, leading to insulin resistance, dyslipidemia, and increased levels of C-reactive protein (CRP), fibrinogen, and plasminogen activator inhibitor-1 (PAI-1), respectively. Reprinted from Libby.24 Copyright © 2008, with permission from Elsevier.
Congestive heart failure also contributes to excess cardiovascular mortality in RA.29 In recent echocardiographic studies, left ventricular systolic dysfunction was three times more common in RA patients compared with the general population.30 Both right and left ventricular diastolic dysfunction has been documented in this disease population despite the lack of clinically evident cardiovascular disease.31 The mechanisms by which RA patients develop heart failure are unclear. Although in some patients congestive heart failure may develop secondary to ischemic heart disease, most cases of heart failure associated with RA are less likely than other patients with heart failure to have a history of ischemic heart disease, obesity, or hypertension, yet they have significantly increased mortality.32
There is currently a lack of evidence-based guidelines for the management and prevention of cardiovascular disease in RA patients. In treating RA, the primary goal is tight control of joint inflammation, which is done using conventional disease modifying antirheumatic agents (eg, methotrexate) as well as biologic therapy (eg, TNFα inhibitors, tocilizumab, abatacept). TNFα inhibitors are generally avoided in RA patients with a history of heart failure because of studies showing increased morbidity and mortality in non-RA patients with congestive heart failure who were treated with a TNFα inhibitor.33 In general, traditional cardiovascular risk factors should be aggressively managed in patients with RA until further studies have evaluated the relative benefits and risks of this approach.33
Adult-onset Still disease refers to a syndrome, usually affecting young adults, that presents with polyarthritis in the setting of fever, evanescent rash, and sore throat (similar to systemic juvenile inflammatory arthritis in children). Pericarditis is the most common cardiac manifestation of adult Still disease, occurring in up to 24% of patients.34 There have been recent case reports, albeit rare, of pericarditis complicated by life-threatening cardiac tamponade.35 Adult onset Still disease is usually treated with corticosteroids and methotrexate, although TNFα inhibitors, such as etanercept and adalimumab, and the IL-1 antagonists, such as anakinra and rilonacept, as well as tocilizumab (anti-IL-6 receptor antibody), appear to be promising alternatives for severe or refractory cases.
Systemic lupus erythematosus (SLE) is a much less common systemic autoimmune disease that primarily affects young women, with peak incidence between the ages of 15 and 40 years. In the United States, its prevalence is estimated to be 1 in 2000 individuals and more commonly affects Hispanics and African Americans than Caucasians.36 Although more than 95% of SLE patients test positive for serum antinuclear antibodies (ANA), not all patients with a positive ANA have SLE. SLE patients are clinically a heterogeneous group, with a wide range of disease severity and spectrum of organ system involvement. Cardiovascular involvement, in particular, occurs commonly in patients with SLE. It is noted in up to 70% of patients by autopsy37-39 as well as by abnormalities seen on echocardiography.40-42
The pathogenesis of SLE is likely to be multifactorial with dependence on genetic risk factors. The mechanisms of disease are complex, with contributions from both aberrantly regulated innate (eg, increased expression of genes stimulated by type 1 interferon) and adaptive immune responses (eg, autoantibodies). However, several lines of evidence support a prominent role for immune complex deposition in disease mechanisms, which may be relevant to the pathogenesis of vasculitis and possibly accelerated atherosclerosis in SLE.
The most common cardiovascular manifestation of SLE is pericarditis,43 with up to 42% of patients in one echocardiographic study showing an effusion.41 Pericardial effusions may be detected at any point in the disease and are usually asymptomatic and small. Acute pericarditis may occur in as many as 20% to 30% of patients with SLE.44 Rarely, acute pericarditis may be associated with cardiac tamponade45; chronic pericarditis may occasionally lead to constriction.46 Particular attention must be paid to differentiating lupus pericarditis from infectious causes in the setting of concomitant immunosuppressive therapy for SLE.44 Treatment of lupus pericarditis depends on its severity. Although no therapy is required for asymptomatic, small effusions, symptomatic, acute pericarditis may warrant treatment with nonsteroidal anti-inflammatories or corticosteroids. Acute lupus pericarditis of moderate severity or worse is usually treated with a course of prednisone beginning at 40 to 60 mg daily followed by a subsequent taper in the dose over several weeks according to the clinical response.
The valvular lesions of SLE are noninfectious verrucous vegetations, which can be found on the ventricular surface of the mitral leaflets; this condition is referred to as Libman-Sacks endocarditis (Fig. 89–2). Although any of the four valves may be affected, valvular abnormalities occur most commonly on the mitral valve, with resultant mitral regurgitation, followed in frequency by the aortic valve.43 Valvular lesions are not uncommon, but usually are asymptomatic. According to one series, more than 50% of patients with SLE had valvular abnormalities by transesophageal echocardiography.47 The most common abnormality in this study was valvular thickening, followed by vegetations, and then valvular insufficiency. The significance of valve thickening is unknown and may resolve over time or worsen. Complications of valvular vegetations included cerebral and coronary artery thromboembolism,48,49 although the absolute risk is very low. Other abnormalities that have been reported in SLE include valvulitis,50 valve fibrosis and mucoid degeneration, and aortic dissection.51
Figure 89–2.
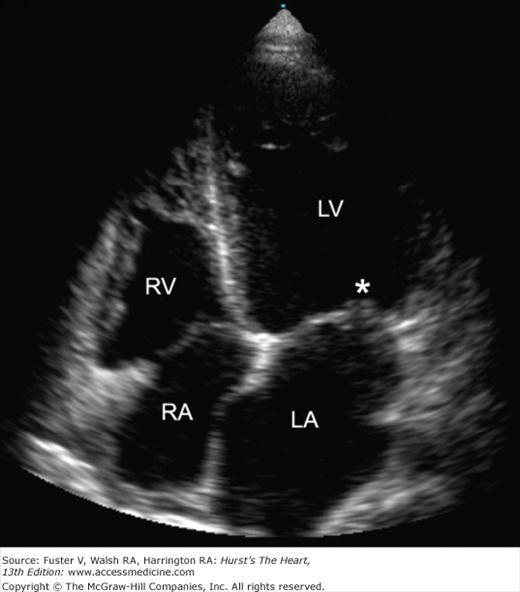
Echocardiographic image of Libman-Sacks endocarditis. Transthoracic echocardiogram (apical four-chamber view) of a patient with systemic lupus erythematosus and Libman-Sacks endocarditis. Note the thickening and vegetation (asterisk) of the mitral valve. LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle.
There is a lack of consensus regarding the use of corticosteroids for treatment of valvular vegetations or other abnormalities. Valve replacement surgery is the usual course of action for the management of patients with clinically symptomatic or hemodynamically significant valve disease.50,52 The American Heart Association guidelines recommend antibiotic prophylaxis in all SLE patients with valvular abnormalities undergoing intestinal, invasive dental, or genitourinary procedures.53
Myocardial dysfunction in SLE may result from valvular disease, coronary artery ischemia, or sustained hypertension and in some cases can lead to clinically significant congestive heart failure. Lupus cardiomyopathy, which is defined as a cardiomyopathy in the absence of ischemic disease or hypertension, has been reported in many series but is not well understood from a pathophysiologic perspective.54 Acute myocarditis should be suspected in any patient with SLE that presents with new-onset arrhythmia, fever, dyspnea, and chest pain. The diagnosis of lupus myocarditis is often based on clinical grounds after evaluation by coronary angiography and other cardiac imaging procedures. Cardiac enzymes are not usually elevated in lupus myocarditis. The role of endomyocardial biopsy is not well established; it is subject to sampling error, and the sensitivity and specificity of the biopsy findings in myocarditis are unknown.53 Treatment of clinically significant myocarditis typically calls for high doses of prednisone therapy for a prolonged course of 3 to 6 months with or without other immunosuppressive agents.
Lupus myocarditis may be complicated by tachyarrhythmias and conduction system disturbances. Additionally, injury to the conduction system, a rare occurrence, may result from small-vessel vasculitis, leading to various forms of heart block. Pericarditis may be associated with atrial fibrillation and flutter, although it is usually transient. Unexplained sinus tachycardia may also occur in SLE patients without obvious cardiac involvement and resolve with steroid therapy.44
A conduction system abnormality may occur in infants of mothers with SLE whose serum contains anti-Ro and anti-La antibodies. Some of these mothers may also have been previously diagnosed with primary Sjögren syndrome, whereas others may appear healthy. Fewer than 5% of women with anti-Ro and/or anti-La antibodies will give birth to infants with neonatal lupus syndrome or congenital heart block. Congenital heart block develops from the transmission of maternal anti-Ro and anti-La antibodies to the fetus, causing myocardial inflammation and fibrosis.55 High-risk mothers with these serologic features should undergo fetal echocardiograms to screen for conduction system abnormalities or evidence of myocardial dysfunction. If any such abnormalities are identified, then treatment with dexamethasone and plasma exchange may be helpful in ameliorating the fetal myocarditis; however, the evidence supporting this approach is anecdotal.56
Coronary artery disease is prevalent in SLE and has emerged as a significant cause of morbidity and mortality for these patients.57 Coronary artery disease may result from coronary arteritis or thrombosis, but is most often secondary to atherosclerosis. Making a diagnosis of coronary arteritis is challenging owing to the nonspecificity of the angiographic findings. It may require sequential angiographic studies showing changes in luminal blockages over time not expected with atherosclerosis. Coronary arteritis, if suspected, may be treated with high doses of corticosteroids, possibly in combination with another potent immunosuppressive agent such as cyclophosphamide. Coronary thrombosis may be associated with the presence of antiphospholipid antibodies (see next section) or embolism from a valvular vegetation, as seen in Libman-Sacks endocarditis.
Recent data suggest that subclinical atherosclerosis is highly prevalent amongst SLE patients.57 Women with SLE between the ages of 35 and 44 years are 50 times more likely to have a myocardial infarction than controls.58 Young women with SLE may have several risk factors for coronary atherosclerosis, such as hypertension, which may be secondary to renal disease and diabetes brought about or worsened by corticosteroid exposure. In epidemiologic studies, SLE has been shown to be an independent risk factor for cardiovascular disease.58 However, the mechanisms underlying this predisposition are unclear, and their relationship to the systemic inflammatory response in SLE remains an area of investigation. Recently, patients with SLE have been shown to produce proinflammatory forms of high-density lipoprotein that confer an increased risk for atherosclerosis.59 Because SLE is predominantly a disease of young women, it is important to recognize that this group is at increased risk for coronary artery disease, and prompt evaluation is warranted if they develop any symptoms of cardiac ischemia.
Drug-induced SLE (DIL) is a rare complication of certain medications, including procainamide, quinidine, and hydralazine. Other medications associated with DIL include isoniazid, minocycline, clindamycin, and phenytoin. DIL, which affects males and females equally, is associated with the development of serum anti-histone antibodies, although only a few will actually develop the clinical syndrome. Common signs and symptoms of DIL include pericarditis, pleuritis, arthralgia, and fever. It is rare to develop renal or neurologic complications from DIL. The syndrome will usually resolve on its own after a short course of prednisone and discontinuation of the offending medication.60
Antiphospholipid syndrome (APS) is a disorder characterized by the clinical triad of recurrent arterial or venous thromboses, pregnancy loss, and thrombocytopenia. Serologically, it is defined by the presence of anticardiolipin antibodies, anti-β2 glycoprotein antibodies, or a positive lupus anticoagulant. APS may occur independently, referred to as primary APS, or be associated with SLE or other autoimmune disease, referred to as secondary APS.
Because APS may produce thrombotic occlusion of many different types and sizes of blood vessels, it can produce a variety of cardiovascular manifestations. Valvular disease is the most common APS-related cardiovascular manifestation. It is seen in both primary and secondary APS and is essentially indistinguishable from Libman-Sacks endocarditis. Coronary artery disease and accelerated atherosclerosis have also been associated with APS, although it seems that all types of antiphospholipid antibodies do not equally contribute to atherosclerosis. In a study by Soltész and colleagues,61 presence of lupus anticoagulant was more frequently associated with venous thrombosis, whereas anticardiolipin antibodies were more often associated with carotid, peripheral, and coronary artery disease. The link between myocardial infarction and APS is less certain. Although some studies have shown a positive correlation between serum antiphospholipid antibodies and myocardial infarction,62-64 other large cohorts have not found this relationship.65,66 Intracardiac thrombi, cardiomyopathy, and pulmonary hypertension have also been associated with APS.67
Dermatomyositis (DM) and polymyositis (PM) are two inflammatory muscle diseases with cardiovascular implications. Patients with DM usually present with skin involvement characterized by erythematous scaliness over the knuckles (Gottron papules), elbows, and knees, as well as periorbital swelling and a violaceous rash around the lids, known as a heliotropic rash. They may also display a photosensitive rash over the face, chest, and back in a shawl-like distribution. Both DM and PM are manifested by proximal muscle weakness and most show increases in serum levels of muscle enzymes such as creatine kinase and aldolase. Although the pathophysiology of DM and PM is incompletely understood, these diseases are associated with a specific set of serum autoantibodies, including antiaminoacyl-tRNA synthetase (antihistidyl-tRNA synthetase and others), antisignal recognition particle, and anti-SNF2 superfamily nuclear helicase (anti-Mi-2). The histopathology of the muscle lesions is different in the two diseases. DM is characterized by a mixed T- and B-cell perivascular infiltrate with perifascicular atrophy. In contrast, muscle biopsies from patients with PM show a predominance of T cells and a diffuse or patchy inflammation of the muscle fascicles. DM may also be associated with a systemic angiopathy. In older patients, DM and PM may evolve as a paraneoplastic syndrome. Although these diseases primarily affect striated muscle, they may also cause cardiovascular complications.
Cardiovascular complications of DM/PM include pericarditis, conduction system abnormalities, congestive heart failure, and myocarditis. Pericardial involvement has been noted most often in patients with overlap syndromes (ie, features of two or more connective tissue diseases). Conduction abnormalities, nonspecific ST-T changes, and left ventricular diastolic dysfunction have also been reported in DM/PM.68 A serious cardiovascular complication is myocardial infarction secondary to coronary vasculitis.69 Myocarditis may infrequently cause heart failure, although newer imaging modalities, particularly cardiac MRI with gadolinium enhancement, may detect subclinical disease in as many as half the patients with DM/PM and be useful in monitoring response to therapy.70
It is important to rule out other forms of myopathy in patients who present with muscle weakness. Patients taking statins may develop a myopathy mimicking inflammatory muscle disease. Statin-related myopathy and inflammatory myopathy may be differentiated by certain aspects of the medical history (eg, statins usually associated with significant myalgia), the results of electromyography, and findings on muscle biopsy, if necessary. For statin-induced myopathy, withdrawal of the offending drug will lead to resolution of the myopathic features. The cornerstone of the treatment for DM/PM is high doses of corticosteroids, usually in doses of 1 mg/kg for several months with a slow taper. Adjunctive therapy often includes methotrexate or azathioprine. Intravenous immunoglobulin may be effective for treating refractory and severe cases.
Systemic sclerosis (or scleroderma) is a rare disorder characterized by microvascular injury and excessive fibrotic changes that can affect multiple organ systems. Most commonly, systemic sclerosis causes hardening of the skin, but can also affect visceral organs, such as lungs, kidneys, and the heart. The diffuse or progressive type of systemic sclerosis results in widespread cutaneous involvement of the distal and proximal extremities as well as the trunk and is usually associated with early and serious visceral involvement. In contrast, limited cutaneous systemic sclerosis, also known as CREST syndrome, is characterized by calcinosis cutis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasias. It may be associated with late involvement of visceral organs, especially increased risk for pulmonary artery hypertension (PAH). Cardiovascular involvement of systemic sclerosis may occur with either the diffuse or limited forms, as well as overlap syndromes in which patients may have features of systemic sclerosis in combination with those of SLE, RA, or polymyositis.
Pericardial disease in systemic sclerosis is usually benign. On the basis of autopsy results, the incidence of pericardial involvement is approximately 50%, but symptomatic pericarditis only manifests in approximately 16% of patients with diffuse scleroderma and in approximately 30% of patients with limited scleroderma.71 Pericardial effusions rarely cause symptoms, although they can be detected in approximately 40% of patients by echocardiography. In most cases, the effusion is relatively small and of no clinical consequence. Routine pericardiocentesis in the absence of cardiac tamponade has no apparent effect on the outcome of these patients.71 Pericardial involvement can present either as acute pericarditis associated with dyspnea, chest pain, and pericardial friction rub, or as a chronic pericardial effusion owing to congestive heart failure. For the treatment of acute pericarditis, nonsteroidal anti-inflammatory therapy is recommended with careful observation of renal function. Pericardiocentesis or surgical intervention is considered only if pericarditis is complicated by tamponade or constriction or if acute infection is suspected. Corticosteroids are generally considered to be of limited benefit, but they may be lifesaving in the setting of associated myocarditis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


