Fig. 12.1
(a) Case 1: Chest x-ray shows that the left diaphragm is obscured by a pleural effusion. The abdomen is protected by a lead shield. (b–d) Case 1: CT chest (selected images) showing that the left hemothorax and left-lower AVM (b, c) and left upper lobe AVM (d)
CT chest (Fig. 12.1b–d) reveals a moderate left sided effusion (suspected hemothorax) with pulmonary arteriovenous malformations (AVMs) in the left lower lobe, left upper lobe and the right lower lobe. Transcatheter embolization of the pulmonary AVMs was performed by an experienced interventional radiologist. The patient went into labour at 38 weeks of pregnancy and gave birth to a healthy child. Chest-x-ray performed in follow-up shows the embolization coils bilaterally (Fig. 12.2). On further history, the patient reports recurrent epistaxis since the age of 12. Family history reveals recurrent spontaneous epistaxis in the father, as well as stroke. The patient, and eventually her family, was thus diagnosed with Hereditary Hemorrhagic Telangiectasia (HHT).
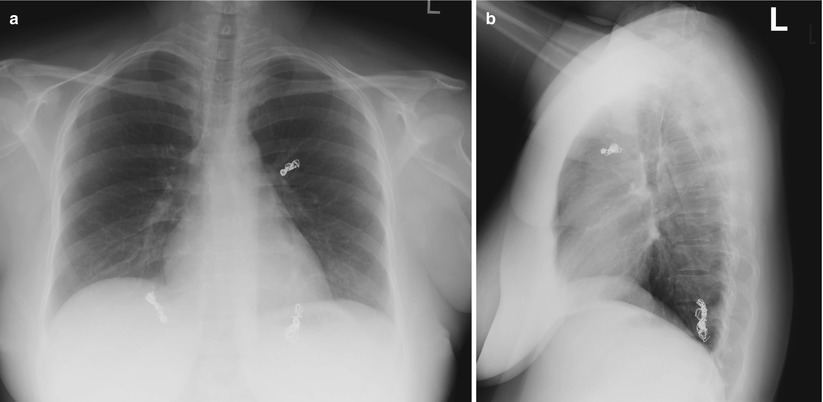
Fig. 12.2
(a, b) Case 1: Chest x-ray (PA and lateral) after embolization shows coils in the left upper lobe, left lower lobe and right lower lobe
Pulmonary Arteriovenous Malformations
Pulmonary AVMs are associated with underlying HHT in more than 80 % of patients [67]. Most other pulmonary AVMs are considered idiopathic [70] but they have also been very rarely reported in association with hepatopulmonary syndrome, schistosomiasis, mitral stenosis, trauma, actinomycosis, Fanconi’s syndrome and metastatic thyroid carcinoma [33]. The detection of pulmonary AVMs, or their complications, may however predate the HHT diagnosis, particularly as HHT it is an under-recognized disorder.
HHT is an autosomal dominant disease, characterized by the presence of vascular malformations (telangiectasia and AVMs) and caused by mutation in either the Endoglin gene or the ACVRL1 gene in 80 % of families. Pulmonary AVMs have a higher prevalence in patients with Endoglin mutation (49–75 %) than in patients with an ACVRL1 mutation (5–44 %) [43, 45, 56]. These genes are involved in transforming growth factor- beta (TGF-ß) signaling pathway and though the disease is characterized by dysregulated angiogenesis, the exact pathophysiologic mechanisms of AVM development remains to be elucidated.
Most (80 %) pulmonary AVMs are simple fistulas consisting of a feeding artery directly connected to a draining vein, with only an intervening aneurysmal sac but no capillaries. About 20 % of pulmonary AVMs are complex with multiple feeding arteries, or have multiple draining veins or a septated aneurysmal sac [66]. A severe diffuse form of pulmonary AVMs is present in approximately 5 % of pulmonary AVM cases [22, 53].
Patients with pulmonary AVMs report exertional dyspnea, though only in approximately 50 % of patients. Less than 10 % present with classical features such as cyanosis, clubbing and pulmonary bruit. More typically, patients present with complications from pulmonary AVMs, such as massive hemorrhage or stroke. Hemorrhagic complications develop due to spontaneous rupture of a pulmonary AVM, leading to massive hemoptysis or hemothorax. This complication has occurred in 3–13 % of patients by the time of diagnosis of pulmonary AVMs. Even more frequently, patients develop neurologic complications, such as stroke, transient ischemic attack or cerebral abscess, with frequencies of 10–60 %, 6–47 % and 8–19 % respectively, by the time of diagnosis of pulmonary AVMs [17, 51, 67]. The presumed mechanism for stroke is via paradoxical embolisation of thrombus from the leg deep venous system or alternatively from in-situ thrombus in the AVM. Cerebral abscess in these patients can be caused by a variety of pathogens, but are most commonly due to pathogens typical of periodontal source [49, 58, 60]. Interestingly, migraine is also frequently reported in HHT patients with pulmonary AVM, particularly with aura [54, 63]. There are multiple mechanistic theories for the connection between migraine and pulmonary AVM, from impaired pulmonary capillary clearance (due to shunting) of vasoactive molecules to recurrent paradoxical emboli through pulmonary AVMs.
Pulmonary AVM complications can be largely prevented, with appropriate screening and preventative therapy. The International HHT Guidelines [25], recommend screening all patients with HHT (or suspected HHT) for pulmonary AVMs, and treating preventatively. The recommended first-line screening test is transthoracic contrast echocardiography, with agitated saline, for the detection of right-to-left shunt. This is a low-risk and minimally invasive screening test, with a high sensitivity (93 %) and an excellent negative predictive value (99 %) for the presence of a pulmonary AVM [18, 65]. When there is evidence of right-to-left shunt on contrast echocardiography, CT chest is the recommended diagnostic test to confirm or rule out the presence of pulmonary AVMs [25], and this can be done without enhancement in most cases.
The degree of shunt on contrast echocardiography can be graded (1–4) according to the opacification of the left ventricle [4]. In case of a grade 1 shunt, there is minimal opacification of the left ventricle. In case of a grade 4 shunt there is extensive opacification of the left ventricle with outlining of the endocardium. The number of cardiac cycles after which contrast appears in the left ventricle is not predictive of an intracardiac or intrapulmonary shunt [72]. Increasing shunt grade is associated with increasing positive predictive value of the presence of AVMs requiring embolization [52, 65, 72].
Preventative transcatheter embolotherapy is recommended, by an experienced interventional radiologist, with the goal to occlude pulmonary AVMs with a feeding artery of 3 mm or greater (and in some cases those between 2 and 3 mm) [25]. Currently there are several devices being used for embolization, including various types of coils and also Amplatzer plugs. The reperfusion rates (mostly secondary to recanalization) with coils and Amplatzer plugs are similar, 7–10 % [44]. Embolization is generally performed as a day procedure, or with overnight admission, under local anesthesia and conscious sedation.
The most common complication of embolization is pleuritic chest pain post-procedure, occurring in up to 30 % of patients. The pain is usually self-limiting, lasting on average 7–10 days, and treated with non-steroidal anti-inflammatory drugs, as needed. Other complications, although rare, include lung infarction, transient hemoptysis (vessel perforation), migration of the device into the systemic circulation, with very rare angina pectoris, TIA, cerebral infarction [44, 48]. A migrating device mostly occurs at the time of device placement and in most cases the device can be retrieved by the interventional radiologist via the catheter, during the same procedure.
Follow-up after embolization is routinely performed 1 month after the procedure with an arterial blood gas (including oxygen shunt testing where available), to document improvement in PaO2 and a chest-x-ray to assess for early involution of the aneurysmal sac and draining vein. Subsequent follow-up is recommended after 1 year with a repeat unenhanced CT chest to confirm involution of the aneurysm and of the draining vein of the embolized AVMs [25]. If there is not sufficient involution, reperfusion is suspected and retreatment should be considered. The second goal on CT is to detect growth of residual small AVMs and the rare development of new AVMs. In the case of a negative CT chest 1 year after embolization, repeat follow-up in the future is recommended after 3 years [25].
For patients with HHT with negative transthoracic contrast echocardiography at baseline, rescreening is recommended every 5 years. Patients who are found to have small pulmonary AVMs on CT chest, with feeding artery <2 mm diameter and not causing complications, can be observed and followed with repeat CT chest every 1–3 year to detect growth and subsequent indication for embolization.
Pulmonary AVM precautions are recommended in all HHT patients with pulmonary AVMs, regardless of treatment, and also all HHT patients with a right-to-left shunt on transthoracic echocardiography, even if there are no CT-detectable AVMs. First, patients should receive prophylactic antibiotics, for all bacteremic procedures, to prevent cerebral abscess and other septic emboli. In addition, dental hygiene should be optimized. The specific choice of antibiotics for prophylaxis depends on the procedure, following the antibiotic choices detailed in the SBE guidelines of the American Heart Association [25, 69]. Secondly, in order to reduce the risk of air embolus, caution should be used to avoid air bubble introduction with intravenous access, preferably by the use of an air-eliminating filter, if available. Finally, it is recommended that patients avoid SCUBA diving to prevent complications from decompression.
Pregnancy and Pulmonary Arteriovenous Malformations
Pregnancy is associated with an increased risk of hemorrhage from pulmonary AVM [58, 60], presumably secondary to the increased cardiac output and increased stroke volume [71]. To reduce this risk, screening for the presence of pulmonary AVMs in HHT patients is recommended prior to pregnancy, with preventative treatment [20]. When pulmonary AVMs are newly diagnosed during pregnancy, embolization is recommended during the second trimester to prevent complications. If pulmonary AVMs are present and not treated during pregnancy, the pregnancy should be considered high-risk. If embolization is performed during pregnancy, it should be performed by an experienced radiologist, with every effort to minimize radiation exposure for the fetus. Exposure reduction can be achieved by covering the abdomen and pelvic area with a lead apron, collimation of the radiation field and limiting of the fluoroscopy time. Taking these precautions will expose the fetus to a radiation dose of <50–200 mrad, which is below the maximal occupational radiation dose for a pregnant worker of 500 mrad [30].
Children with Hereditary Hemorrhagic Telangiectasia
Children with HHT should be screened for pulmonary AVMs as well [19, 25]. Twenty-three percent of asymptomatic children with a HHT diagnosis have pulmonary AVMs, of these 70 % with a significant feeding artery diameter of ≥3 mm [2]. Initial screening for pulmonary AVMs in the pediatric population can be done by upright and supine pulse oximetry, chest radiography and/or transthoracic echocardiography [3, 25]. When screening is positive, CT chest is recommended as it is in adults, to confirm the presence of pulmonary AVMs and measure the feeding artery diameter. Embolization is recommended in children who are symptomatic of the pulmonary AVMs or who are hypoxemic. Treatment of asymptomatic children should be considered on a case by case basis [25]. Treatment by transcatheter embolotherapy in children is low-risk in experienced hands, with complication rates comparable to those in adults [23].
Reports of pulmonary AVMs in neonates are rarer. There are 18 case reports of neonates with pulmonary AVMs, 39 % died within the first week. We suspect there is a reporting bias here, with primarily severe cases being identified and reported at birth. Embolization can be performed in neonates, as in children.
Diffuse Pulmonary Arteriovenous Malformations
Diffuse pulmonary AVMs are pulmonary AVMs occurring in every subsegmental artery of one or more pulmonary lobes. They occur in 4.4 % of patients with pulmonary AVMs and these patients more frequently present with cyanosis, hemoptysis and/or neurologic complications. Most (81 %) of patients with diffuse pulmonary have HHT. Patients can have unilateral or bilateral diffuse pulmonary AVMs, the latter occurring in the majority of patients (72 %), mostly affecting women. Usually patients present at a young age, mean 24 years old, with cyanosis. The majority of patients (70 %) have had neurologic complications by the time of diagnosis.
The mean PaO2 at presentation is 47 mmHg and 75 % of patients have polycythemia due to chronic hypoxemia. Diffuse pulmonary AVMs are associated with an increased mortality of 25 % during a mean follow-up of 8.5 years, only reported in patients with bilateral involvement [22]. Death was due to pulmonary hemorrhage, cerebral abscess or complications from other organ involvement from HHT. Treatment for diffuse pulmonary AVMs is similar to patients with focal pulmonary AVMs, with preventative embolization of AVMs with feeding artery diameter of ≥3 mm. Post-embolization, the PaO2 improves in patients with unilateral involvement, but not significantly in most patients with diffuse involvement.
Patients with HHT can be affected by other vascular malformations (VMs) besides pulmonary AVMs. Most common locations for other VMs are the brain and liver. Though there is no international consensus on asymptomatic screening for brain AVMs, this is the current standard of care in HHT Centres of Excellence across North America, using MRI. Diagnostic testing for the liver VMs is generally only recommended in symptomatic patients, since preventative treatment is not recommended [25], or in cases where the documentation of liver VMs might help complete the clinical criteria, in a given patient, for diagnosis of HHT.
Clinical vignette 2
A 70 year old woman presents with progressive exertional dyspnea and ankle edema. On physical examination her jugular venous pressure is elevated, she has mucocutaneous telangiectasia and she has ascites. An electrocardiogram reveals atrial fibrillation. Transthoracic echocardiography reveals an elevated estimated right ventricular systolic pressure of 43 mmHg, suggestive of pulmonary hypertension. Doppler ultrasound of the liver reveals a dilated hepatic artery at 7.5 mm with an increased hepatic artery peak flow velocity (120 cm/s) and decreased resistive index (0.55). Multi-detector triphasic helical CT reveals diffuse liver VMs with arterioportal shunt.
Pulmonary Hypertension
Pulmonary hypertension (PH) refers to an increased pulmonary arterial pressure, which can subsequently lead to right heart failure. Patients with HHT can present with PH, most commonly secondary to the presence of liver VMs (class 2 pulmonary hypertension), or patients can develop pulmonary arterial hypertension (PAH) (class 1 pulmonary hypertension) [61]. Patients with PH secondary to liver VMs mostly present with an increased cardiac output, while patients with class 1 PAH usually have an elevated mean pulmonary artery pressure and increased pulmonary vascular resistance [24, 29].
Pulmonary Hypertension Secondary to Liver Vascular Malformations
Liver VMs are highly prevalent in HHT, associated with all genotypes, though more frequent in patients with ACVRL1 mutation (84 %) versus patients with an Endoglin mutation (60 %) [56]. Only 5–8 % of patients with liver VMs are symptomatic, based on cross-sectional studies [9, 62]. Liver VMs are more prevalent in HHT patients over 40 years of age [8] and women appear to be more frequently affected than men [10]. Liver VMs with severe shunting can eventually lead to high output cardiac failure (HOCF), typically in the sixth or seventh decades of life. Rarely, women can also present with HOCF from liver VMs during pregnancy [35, 47].
HOCF develops secondary to arteriovenous (hepatic artery to hepatic vein) shunt and/or portosystemic (portal vein to hepatic vein) shunt. Arterioportal (hepatic artery to portal vein) shunts more typically lead to portal hypertension, ascites and esophageal varices. There is often evidence of mixed shunt in symptomatic patients. Arteriovenous shunting leads to a hyperdynamic circulatory state. Subsequently, increased left atrial pressures and impaired pulmonary vasodilatation cause PH. PH and volume overload will lead to right ventricle strain and eventually dilatation, which subsequently lead to right ventricle enlargement and contractile dysfunction, leading to tricuspid regurgitation and eventually right heart failure [24].
Patients with PH secondary to liver VMs typically present with symptoms of HOCF: fatigue, palpitations, exertional dyspnea, orthopnea and peripheral edema. On physical examination a triad can be found of wide arterial pulse pressure, systolic ejection murmur at the left sternal border due to tricuspid regurgitation and a hepatic bruit.
Liver VMs can be detected by Doppler ultrasound of the liver. Major findings on Doppler ultrasound in these patients are hepatic artery dilatation (>0.7 cm) and intrahepatic arterial hypervascularization. Minor criteria are the presence of increased hepatic peak velocity >110 cm/s, decreased hepatic artery resistance index <0.6, an increased portal vein peak velocity >25 cm/s and the tortuous course of the extrahepatic artery [13]. Triphasic hepatic CT, MRI or mesenteric angiography are options for diagnostic confirmation of liver VMs and also provide more detailed information reading the type(s) of shunting present as well as other complications (biliary cystic dilatation, focal nodular hyperplasia, etc.) [25].
The suspicion of PH and HOCF is generally confirmed on transthoracic echocardiography, but right heart catheterization is helpful in cases where the association with liver VMs, or the cause of PH, is uncertain. Patients with PH secondary to liver VMs will have elevated mean pulmonary artery pressure, markedly increased cardiac output, normal pulmonary vascular resistance, normal transpulmonary gradient and elevated pulmonary capillary wedge pressure [31].
Routine management of HOCF due to liver VMs includes salt restriction, diuretics, beta-blockade and treatment of anemia and atrial fibrillation [11].
Bevacizumab can be considered in refractory cases, and/or liver transplantation. Though experience with this antibody against vascular endothelial growth factor (VEGF) is limited, results have suggested it may have a role in these refractory cases, and transplantation may be avoided. Interestingly bevacizumab has been shown to improve the cardiac output in patients with liver VM and high cardiac output, with a complete response in 22 % of patients and a partial response in 65 % of patients at 6 months follow-up. A complete response is considered a normalization of the cardiac index, which should be 2.5–3.9 L/min/m2 in men and 2.5–3.6 L/min/m2 in women. Bevacizumab treatment also reduced the mean duration of epistaxis and improved quality of life. Treatment had no effect on hepatic artery diameter or peak flow velocity [21].
Liver transplantation is considered in refractory liver VM patients, for HOCF, portal hypertension or biliary necrosis. Perioperative mortality of liver transplantation in patients with HHT is reported at 10–17 %, due to hemorrhage (intraoperative, cerebral, pulmonary, gastric), heart failure or rejection of the liver or primary nonfunctional liver. After liver transplantation the cardiac function improved in 75 % of patients and stabilized in 21 % of patients. The 10 year survival rate after liver transplantation in patients with HHT is 83 % [42]. Patients with increased cardiac output and normal peripheral vascular resistance and normal right ventricle function are eligible for liver transplantation. Patients with severe pulmonary hypertension (mean pulmonary artery pressure ≥35 mmHg), elevated pulmonary vascular resistance (≥250 dyn·s·cm−5) and right ventricle dysfunction unfortunately are considered higher risk as liver transplantation in these patients is associated with increased mortality [26].
Surgical hepatic ligation and percutaneous hepatic artery embolization have been performed to treat the intrahepatic shunt. These procedures carry a significant risk of developing biliary ischemia and/or hepatic necrosis. In view of these serious complications, these procedures are generally not recommended, though are occasionally considered for patients with refractory disease who are not considered candidates for liver transplantation [12, 42].
Pulmonary Arterial Hypertension
Familial PAH is a rare disorder, with an estimated prevalence of 15 per million [37] and rarely caused by HHT. Within HHT patients, the prevalence of pulmonary arterial hypertension is not known.
PAH also occurs in HHT patients, though rarely (approximately 1 % of HHT patients). Most affected to date have ACVRL1 mutation [32, 36, 64]. PAH can be suspected based on symptoms: dyspnea, syncope, fatigue, edema. HHT patients with PAH present with similar symptoms as patients with idiopathic PAH. Pathologic characteristics of arteriopathy in patients with HHT PAH consist of intimal proliferation, medial hypertrophy, plexiform lesions and in situ thrombosis.
The diagnosis is suspected based on symptoms or if an elevated estimated right ventricular systolic pressure (>40 mmHg) is found on routine transthoracic echocardiography. PAH should be confirmed by right heart catheterization, as in patients with idiopathic PAH. Hemodynamic criteria for the diagnosis of PAH are the presence of an elevated mean pulmonary arterial pressure of >25 mmHg in rest, with a normal left atrial or wedge pressure (≤15 mmHg) and increased pulmonary vascular resistance.
Complications from PAH in patients with HHT can be precipitated by anemia, leading to right heart failure. Patients with HHT are at risk for hemorrhage, most commonly from epistaxis or from telangiectasis in the gastrointestinal tract. Severe hemorrhage can lead to hypovolemia or anemia. Hypovolemia leads to a reduced cardiac output and anemia will lead to decreased oxygen delivery which both can contribute to worsening right ventricular failure.
Right heart failure can also be precipitated by embolization of pulmonary AVMs in patients with pulmonary arterial hypertension. Closing the shunt in the pulmonary AVM can lead to increased mean pulmonary artery pressures, with subsequent increase in the right ventricle afterload. However, the risk of massive hemorrhage from untreated AVMs is likely greater in patients with PAH, and therefore decisions about embolization of pulmonary AVMs in the patients must be made on a case-by-case basis.
Management of PAH in patients with HHT is similar to management of patients with idiopathic PAH. Pulmonary vasodilators, i.e. bosentan, an endothelin receptor antagonist, have been reported to have a beneficial effect in patients with HHT and pulmonary arterial hypertension [7, 15], though there is limited evidence. Caution is warranted with the use of pulmonary vasodilators, since systemic vasodilatation could increase any systemic shunt and worsen heart failure. Sildenafil, a phosphodiesterase-5 inhibitor has also been reported to be beneficial in one case of PAH in HHT [14]. Anticoagulation is not absolutely contraindicated in HHT patients, but its use, rather, should be decided on a case by case basis.
Background HHT
HHT is an autosomal dominant inherited disease affecting 1 in 5,000–10,000 persons, characterized by the presence of vascular malformations. HHT has also been previously referred to as Osler-Weber-Rendu disease. HHT is characterized by the presence of mucocutaneous telangiectasia (Fig. 12.3), recurrent epistaxis, visceral AVMs and a positive family history. These characteristics are the four diagnostic criteria for HHT (Table 12.1). The diagnosis of HHT is definite if patients meet ≥3 criteria. The diagnosis is possible if they meet 2 criteria and the diagnosis of HHT is unlikely if patients meet ≤1 criterion [59].