1. Presence of liver disease
(a) Cirrhotic or non-cirrhotic portal hypertension
2. Intrapulmonary vascular dilatations
(a) Positive transthoracic contrast/bubble echocardiogram
(b) 99mTechnetium lung/brain perfusion scan
3. Hypoxemia
(a) Seated room air PaO2 <70 mmHg
(b) Alveolar-arterial gradient on room air >20 mmHg
Prevalence rates for HPS in the literature have ranged from 5 to 29 % in patients with end-stage liver disease undergoing liver transplantation evaluation [2]. The true prevalence of HPS in patients with various stages of liver disease is unknown. In a large prospective study of 80 patients with cirrhosis undergoing liver transplant evaluation, the prevalence rate of HPS was 17.5 % [3].
Positive contrast echocardiography consistent with intrapulmonary shunt with normal gas exchange has been documented in patients with cirrhosis. Table 13.1 compares several studies of patients with cirrhosis evaluated using both contrast echocardiography and arterial oxygenation assessment. Positive contrast echocardiography documenting intrapulmonary vascular dilatation ranged from 3.0 to 20.0 % [3–6]. Arterial hypoxemia ranged from 0.0 to 17.5 %. It is conceivable that patients with positive contrast echocardiography consistent with intrapulmonary shunt, but normal gas exchange, have a higher incidence of developing HPS or have a “preclinical” form of HPS. These patients deserve prospective follow-up to determine their actual outcome.
Table 13.1
The prevalence of positive transthoracic echocardiography in patients with portal hypertension in relation to abnormal oxygenation (true HPS)
N | + CE | + CE | DLCO <80 % | |
|---|---|---|---|---|
− hypoxemia | + hypoxemia | |||
Martinez et al. [3] | 80 | 2 (3) | 14 (17.5) | 14 (18) |
Mimidis et al. [4] | 75 | 8 (10.7) | 0 | 8 (11) |
Gupta et al. [5] | 54 | 4 (7.4) | 6 (11.1) | 5 (9) |
Abrams et al. [6] | 40 | 8 (20) | 7 (17.5) | 11 (14) |
HPS occurs with equal frequency in men and women and, also occurs in pediatric patients [7]. Portal hypertension appears to be a necessary component in the development of HPS [1]. The incidence of HPS is not related to any specific etiology of liver disease [1, 2, 6, 8]. Hypoxemia severity does not correlate with the severity of portal hypertension as measured by portal pressure or the severity of liver disease as measured by the Child-Pugh or MELD scores [6, 8]. Severity of hepatic dysfunction associated with portal hypertension does not predict the presence or severity of HPS. HPS has been documented in patients with mild degrees of hepatic dysfunction (Childs-Pugh “A”) and hypoxemia has progressed in the setting of stable hepatic dysfunction.
Pathophysiology
Gas exchange is the primary abnormality in HPS resulting in hypoxemia caused by pulmonary vascular dilations. A vascular imbalance between vasodilating and vasoconstricting mediators has been proposed as a probable cause for the pathologic changes seen at autopsy [2]. Normal capillary diameter in the pulmonary vascular system ranges from 8 to 10 μm [1]. Both autopsy studies and pulmonary angiography have demonstrated two different types of vascular dilatations in HPS; Types I and II (Fig. 13.1a, b). Remarkably, physiologic studies suggest that these changes can reverse following liver transplantation in most cases [9].
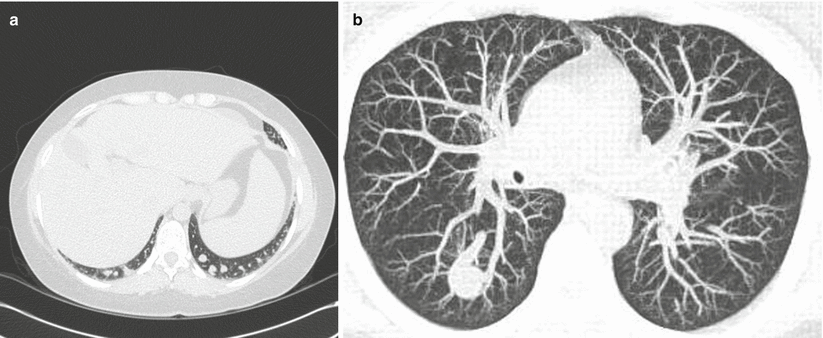
Fig. 13.1
(a) Type I dilatations consist of diffuse pre-capillary and capillary vascular dilatations between 15 and 500 μm in diameter. (b) Type II dilatations involve distinct arteriovenous communications
Type I dilatations consist of diffuse pre-capillary and capillary vascular dilatations between 15 and 500 μm in diameter [2, 10]. These types of dilatations lead to ventilation-perfusion mismatching caused by excessive perfusion for a given unit of normal ventilation. Type II dilatations involve distinct arteriovenous communications resulting in the absence of ventilation with increased perfusion and the presence of true anatomic shunting. The response to 100 % oxygen varies with both the degree and type of intrapulmonary vascular dilatations present [8]. With type I lesions, the response to 100 % oxygen may be near normal, while in a pure shunt (type II lesions), there will be minimal or no response to 100 % oxygen. Dilated pleural vessels similar to spider angiomata in liver disease can also be found in HPS, but they do not appear to participate in gas exchange [10].
The mechanism of hypoxemia in HPS is likely multi-factorial and, in a given patient, may include the co-existence of other non-vascular reasons for hypoxemia [3, 8]. Patients with advanced liver disease often have pre-existing gas exchange abnormalities due to the presence of pleural effusions (“hepatic hydrothorax”), ascites, interstitial lung diseases, and/or obstructive lung disease (emphysema, bronchitis, or asthma). In the absence of these co-existing, “intrinsic” pulmonary abnormalities, at least three mechanisms of hypoxemia documented by the multiple inert gas elimination technique exist in HPS and are described in Table 13.2 [1, 2, 10]. First, due to the hyperdynamic circulation associated with advanced liver disease, the increased cardiac output decreases the transit time through the pulmonary circulation decreasing the time for capillary oxygenation resulting in ventilation-perfusion mismatching (normal ventilation with excessive perfusion).
Table 13.2
Mechanisms of hypoxemia in hepatopulmonary syndrome are shown
Abnormality | Mechanism |
|---|---|
1. Hyperdynamic circulation | Ventilation-perfusion mismatch |
↑ Cardiac output = ↓ transit time in | Normal V/↑ Q |
Capillary bed ↓ time for gas exchange (oxygenation) | |
2. Diffusion – perfusion limitation | Ventilation-perfusion mismatch |
(Type I intrapulmonary vascular dilatations) | Normal V/↑ Q |
(a) ↑ distance to center of capillary = limitation to diffusion | Diffusion abnormality |
3. True right to left shunt | Ventilation-perfusion mismatch |
(Type II intrapulmonary vascular dilatations) | Shunt = absent V/↑ Q |
4. Intrinsic cardiopulmonary disease if present |
A second mechanism of hypoxemia relates to a diffusion-perfusion limitation due to the presence of Type I diffuse intrapulmonary vascular dilatations. As the degree of dilatation increases, distance from the alveoli to the center of the capillary increases and passive diffusion of oxygen molecules is limited. With a higher partial pressure of alveolar oxygen (obtained while breathing 100 % oxygen), there is an increased driving force for oxygen to diffuse into the capillaries, resulting in a dramatic increase in PaO2 despite severe hypoxemia breathing room air [8]. Such diffusion limitation in combination with the possible increased blood transit time from the hyperdynamic circulation seen in liver disease can result in severe arterial hypoxemia at rest (PaO2 <50 mmHg).
The third mechanism of hypoxemia is the presence of right to left anatomic intrapulmonary shunting. This is uncommon as demonstrated by pulmonary angiography or contrast enhanced CT scanning of the chest. Unoxygenated venous blood simply bypasses gas exchange units via direct arteriovenous communications of varying size and configuration. If large (>5 mm in diameter), they may be amenable to coil embolotherapy. These abnormalities may result in severe hypoxemia with very poor response to 100 % inspired oxygen (PaO2 <300 mmHg) [8].
The interaction of vascular mediators between the liver and the lung has been a subject of much interest. Is the diseased liver tilting the balance between vasodilating and vasoconstricting substances so that there is an unopposed action of vasodilators? Does the pulmonary vascular bed require vasomodulating factors produced by the liver to maintain control of the pulmonary circulation? An intriguing response to this question may be found in the pediatric congenital heart disease literature. Pulmonary vascular dilatation with severe hypoxemia develops after the creation of cavopulmonary shunts as a treatment approach to tricuspid atresia (and its associated vascular abnormalities) [11]. In patients who had aberrant hepatic venous drainage (emptying into the left atrium), arterial hypoxemia and intrapulmonary vascular dilatations resolved with redirection of the hepatic venous effluent into the pulmonary arterial circulation. This well-documented experience provides evidence for a probable vascular mediator arising from the liver that affects the structure and vasomotor tone of the pulmonary circulation.
In a bile duct ligation rat model of HPS, pulmonary vascular endothelial nitric oxide synthase levels are increased resulting in a rise in nitric oxide that correlates with the development of pulmonary vasodilatation [12]. Nitric oxide is a potent vasodilator. In a subsequent study by the same investigators, both hepatic and plasma endothelin-1 levels were increased after bile duct ligation in the rat with subsequent portal hypertension [13]. Endothelin-1 is a potent vasoconstrictor. In this situation, endothelin-1 likely acts on the endothelin-B receptors on the endothelial cell to stimulate nitric oxide production and endothelin-1 clearance. The plasma endothelin-1 levels positively correlated with pulmonary endothelial nitric oxide synthase levels and with alveolar-arterial oxygen gradients. No human studies have addressed the endothelin-B hypothesis to date. Indeed, increased levels of exhaled nitric oxide have been documented in HPS at levels (ppb) greater than those measured in cirrhotic patients without hypoxemia [2, 10].
One non-invasive form of evaluating airway mediators includes the measurement of breath markers. Exhaled nitric oxide levels are increased in HPS patients and decline after liver transplantation [14–17]. Extended nitric oxide analysis rather than single breath exhalation flow rates may be helpful in differentiating the actual production site of NO within the respiratory system i.e. alveolar versus airway production of NO [18].
Tumor necrosis factor alpha (TNFα) levels are increased in the common bile-duct ligation rat model of HPS presumably from bacterial translocation resulting in endotoxemia stimulating production of TNFα [19–22]. TNFα further increases NO levels. Pentoxifylline has been shown to improve HPS by blocking TNFα however additional adverse reactions limited its further evaluation [22, 23]. More recently, Liu and colleagues have shown that a specific monoclonal antibody to TNFα improves HPS in cirrhotic rats mediated through the inhibition of TNFα PI13/Akt-NO pathway [24]. Figure 13.2 illustrates the delicate interaction of the various mediators between the liver and the lung.
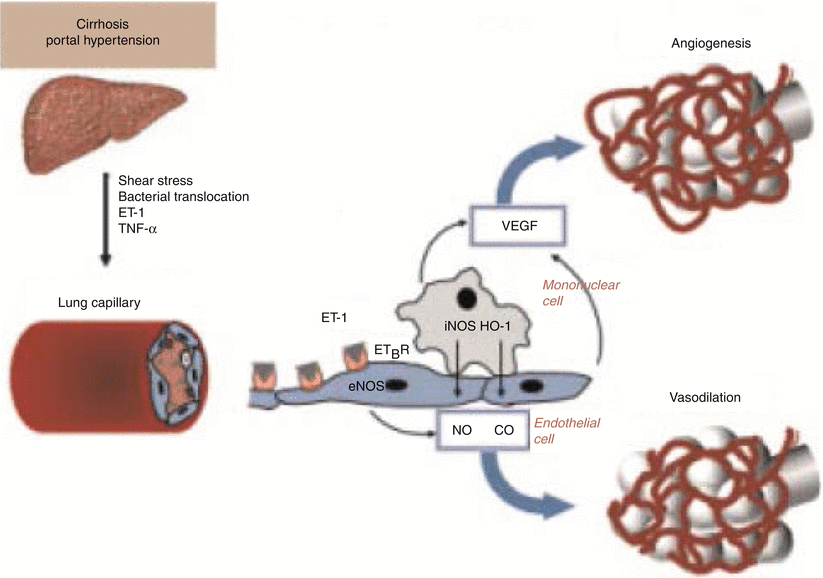
Fig. 13.2
Pathophysiology of hepatopulmonary syndrome. ET-1 endothelin 1, TNFα tumor necrosis factor α, ET B R endothelin B receptor, eNOS endothelial nitric oxide synthase, iNOS inducible nitric oxide synthase (With permission from Machicao and Fallon [50], Thieme Publishers)
In addition to the research on various mediators involved in HPS, genetic risk factors for HPS in patients with advanced liver disease have also been assessed. In a multicenter case-control study of patients with cirrhosis being evaluated for liver transplant, 59 cases of HPS were compared with 126 control patients without HPS [25]. Forty-two single nucleotide polymorphisms in 21 genes were significantly associated with HPS after adjustments for race and smoking. The authors concluded that common genetic variation influences the pathogenesis of HPS and that future studies focusing on genetic patterns are necessary.
Clinical Vignette
A 14 year old female was referred to pulmonary clinic complaining of progressive dyspnea on exertion over the last 12 months; WHO class III functional status. She had developed fatigue and was only able to participate in the first and last periods of school classes per day due to profound dyspnea. Upon presentation, room air arterial blood gas showed the partial pressure of oxygen to be 46 mmHg with an oxygen saturation of 82 %. Pulmonary function studies were normal with the exception of a reduction in diffusion capacity.
On physical examination, cyanosis and clubbing were present. The lung examination was normal and the cardiac examination showed a hyperdynamic state with a grade two systolic murmur present. Mild splenomegaly was identified and there were no hepatic or abdominal bruits.
Computed tomographic chest scan with contrast showed mild cardiomegaly and a prominent spleen with normal lung parenchyma and no evidence of pulmonary embolus. Transthoracic bubble echocardiogram documented a large right to left intrapulmonary shunt. There was no evidence of pulmonary hypertension or valvular heart disease. Nuclear medicine lung-brain perfusion scanning showed a shunt fraction of 66 %. Right and left heart catheterization was performed. Mean pulmonary artery pressure was 31 mmHg with a pulmonary capillary wedge pressure of 17 mmHg and a cardiac output by thermodilution of 9.48 l/min (confirmed by Fick technique). Transjugular liver biopsy was unsuccessful due to chronic stenosis and occlusion involving the hepatic vein confirmed by MR angiography.
Percutaneous liver biopsy was essentially unrevealing. Alkaline phosphatase was 191; AST 82; ALT 58; bilirubin 1.5; GGT 78; ammonia 39; and ferritin 372. Contrast computed tomographic abdominal scan revealed evidence of portal hypertension with a nodular contour to the liver compatible with mild sclerosis; an unusual perfusion pattern; numerous mesenteric varices including paraduodenal veins, varices within Morrison’s pouch, peripancreatic and portal systemic collaterals anteriorly within the abdomen; and a recanalized umbilical vein.
The clinical diagnosis of HPS was established based on the findings of portal hypertension, arterial hypoxemia, and intrapulmonary shunting. She also had an elevation in mean pulmonary artery pressure and pulmonary capillary wedge pressure with high cardiac output resulting in a hyperdynamic state with post-capillary pulmonary hypertension. She underwent an orthotopic liver transplant. Pathology of the patient’s diseased liver showed nodular regenerative hyperplasia with focal septal fibrosis. The patient was discharged on 10 l oxygen after 26 days. Nine months later, the patient was off supplemental oxygen with a normal overnight oximetry; oxygen saturation on room air of 95 %; and lung-brain perfusion shunt fraction of 12 %. Cardiopulmonary hemodynamics normalized.
Clinical Features
The clinical features in HPS are those typically seen in underlying liver disease. On physical examination these include spider angiomata, palmar erythema, gynecomastia, splenomegaly, and jaundice. As the severity of HPS increases, other physical findings may evolve, including cyanosis, tachypnea, clubbing, and orthodeoxia. Orthodeoxia is defined as a decrease in arterial PO2 greater than 3 mmHg in the upright versus the supine position [10, 26]. Patients frequently present with the insidious onset of dyspnea and platypnea (dyspnea exaggerated in the upright position). Orthodeoxia and platypnea although uncommon, are two of the cardinal manifestations of HPS although are not specific for HPS. Worsening hypoxemia in the upright position is due to the increased presence of intrapulmonary vascular dilatations in the mid and basilar portions of the lungs. In the upright position, there is increased perfusion to these dependent areas with worsening ventilation-perfusion mismatching and hypoxemia. When compared to patients with cirrhosis alone, HPS patients have higher incidences of finger clubbing, spider angiomas, and dyspnea [2]. Progressive hypoxemia may occur in the setting of apparently stable hepatic dysfunction [27].
Diagnosis
The diagnosis of HPS is made in the setting of arterial hypoxemia with intrapulmonary vascular dilatation in the setting of chronic liver disease [1]. The hypoxemia in HPS is defined as an arterial PO2 of less than 70 mmHg or an alveolar-arterial oxygen gradient of greater than 20 mmHg (some authors use 15 mmHg) [1, 2]. Arterial blood gases obtained in both the supine and upright position will document the presence and severity of orthodeoxia. Similarly arterial blood gases on room air and 100 % oxygen will document the presence or absence of a true shunt by the response to oxygen. The shunt fraction can be estimated by the 100 % oxygen technique however is based on several assumptions and, in the hyperdynamic state generally present in chronic liver disease, can result in the shunt equation underestimating the true shunt [28].
Two-dimensional transthoracic contrast echocardiography is the most sensitive test to detect the presence of right to left shunting [6]. It is not able to differentiate between the two different types of intrapulmonary vascular dilatations, however can differentiate between intrapulmonary and intracardiac sources of right to left shunt. Contrast echocardiography detects vascular dilatation via micro-bubbles (diameter 10–60 μm) created by hand-agitated saline injected into a peripheral arm vein. Normally these micro-bubbles are absorbed at the capillary-alveolar level through the first pass into the pulmonary circulation. If vascular dilatations exist, the micro-bubbles pass through the pulmonary vascular bed into the left atrium where they can be seen three to five cardiac cycles after the appearance in the right ventricle. If micro-bubbles are seen in the left heart within three cardiac cycles, then an intracardiac shunt may exist (atrial or ventricular septal defect). Helpful indirect echocardiographic markers of HPS compared to non-HPS cirrhotic patients also include left ventricular enlargement and higher systolic velocity in the mitral valve [29].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


