Folliculin gene (FLCN)-associated syndrome
Birt-Hogg-Dubé syndrome
Familial spontaneous pneumothorax
Lymphangioleiomyomatosis
Sporadic
Tuberous sclerosis complex associated
Pulmonary Langerhans cell histiocytosis
Lymphoid disorders
Lymphoid interstitial pneumonia, idiopathic or associated with connective tissue disease (especially Sjögren syndrome)
Pulmonary lymphoma
Amyloidosis
Non-amyloid immunoglobulin deposition disease
Infections
Staphylococcal and other bacterial causes
Pneumocystis jiroveci infection in AIDS and nonAIDS immunocompromised patients
Recurrent respiratory papillomatosis
Hyper-IgE syndrome
Tumours
Metastases of sarcoma
Metastases of benign uterine leiomyoma
Metastases of other tumours
Congenital cystic disorders
Other causes
Genetic disorders (neurofibromatosis, Ehlers-Danlos syndrome, Proteus syndrome, others)
Interstitial lung diseases (hypersensitivity pneumonias, desquamative interstitial pneumonia)
Others (posttraumatic pseudocysts, Erdheim-Chester disease, fire-eater’s lung, etc.)
FLCN Gene-Associated Syndrome (Birt-Hogg-Dubé)
History of Cutaneous, Pulmonary, and Renal Manifestations
Cutaneous Manifestations
Perifollicular fibromas (about 100) which had appeared 6 years earlier on the neck and the nape of the neck were reported in 1925 by R. Burnier and J. Rejsek in a 56 year old woman [4]. Multiple perifollicular fibromas of the face and neck which had appeared in a 53 year old man by the age of 32 years, the brother and a cousin of whom had the same dermatosis (at a lesser degree) were later reported by J. Civatte and J.P. Le Tréguilly [5]. A.R. Birt, G.R. Hogg, and J. Dubé reported in 1977 multiple fibrofolliculomas in a family with hereditary medullary carcinoma of the thyroid, a condition now recognized as a tumour suppressor gene syndrome caused by a germline RET mutation [6]. Thirty-seven members of the kindred of 70 were more than 25 years of age, and 15 of them had fibrofolliculomas (confirmed by biopsy in ten lesions taken from six patients). Trichodiscomas and acrochordons were also present.
Pulmonary Manifestations (Pneumothorax, Lung Cysts)
It is an unusual event which revealed the probable first reported case of the syndrome with pulmonary manifestations in 1960 [7]. JRW, a 20 year-old man, was taught for submarine escape training at US Submarine Escape Training tanks. The technique for escape was as follows. The person escaping enters a special isolated compartment, which is then flooded from the sea to about chest level. With compressed air the pressure within the compartment is increased to equal the outside sea pressure. The escape hatch can then be opened, allowing access to the sea. With an inflated Mae West life jacket the man escaping then steps into the sea and rises to the surface at a rate of about 375 ft per minute. The compressed air within the life jacket is vented through special valves to prevent rupture of the jacket through expansion of its contained air as the surface is approached. The man escaping exhales continually during his ascent.
JRW had a normal chest X-ray. He followed the procedure and left the area. He was found lying on the grass in a stuporous condition 30 mn later, then improved. He did two consecutive other procedures and collapsed after the second. Recompression was done, resulting in improvement, then decompression could be done.
Chest X-ray showed several cystic areas in the left upper lobe, some of which with air levels. It was supposed that this was a case of air embolism associated with acute development of pulmonary cysts during decompression from depth.
Over the 50 following years JRW was admitted five times at hospital for pneumothorax. He further had a curative resection for clear cell-type renal carcinoma. An (astute) intern at hospital eventually considered the possibility of the syndrome. The patient had indeed removal of acrochordons several times. He further had a colorectal adenoma [8].
“Unilateral lung cysts” (not otherwise precised) were reported in 1975 by Hornstein and Knickenberg [9] in a man with further kidney cysts and solid nodules on his face, neck, and back. His daughter had innumerable perifollicular fibromas on the face, neck and trunk, a large goitre, and adenomatous colon polyps one of them transformed into carcinoma.
Perifollicular fibromas were further reported in 1986 in a man with colon polyposis who developed iterative spontaneous pneumothoraces. Spontaneous pneumothorax had occurred in seven people in the family, including four with perifollicular fibromas with further colon polyposis [10].
Chung et al. [11] later reported multiple spontaneous pneumothoraces starting at 15 years of age with bullous emphysema in his early twenties in a male patient with skin fibrofolliculomas.
Toro et al. [12] reported pulmonary cysts present in 4 of 28 patients from kindreds with familial renal tumors. Of these patients 3 had characteristic skin features, and one developed pneumothorax.
Renal Tumours
Bilateral chromophobe renal adenocarcinoma was reported in 1993 by Roth et al. [13] in a patient with characteristic fibrofolliculomas on the face, neck, and upper chest.
Toro et al. [12] in a study of kindreds with familial renal cancer found 13 patients with the syndrome. Of these 3 had lung cysts.
Towards a Novel Non-eponymous Terminology of the Syndrome
The above review clearly demonstrates that A.R. Birt, G.R. Hogg, and W.J. Dubé [6] were not the first to describe familial multiple skin fibrofolliculomas. They further did not describe the other cardinal features of the condition i.e. MCLD and renal cancer.
It has been suggested that the name of Hornstein is added to those of the above authors [14] because he reported multiple perifollicular fibromas, polyps of the colon, and unilateral lung cysts. Further Furaya and Nakatoni [15] suggested to rename the syndrome as the name of the woman physician who worked with Hornstein was omitted. Thus the eponymous name of the condition would become at least Hornstein-Knickenberg-Birt-Hogg-Dubé syndrome, a bizarre terminology.
Because eponyms do not always correspond to the first and/or principal contributors to the genuine entity, we further agree that a descriptive anonymous terminology is more appropriate [16].
We have proposed at the Fifth Birt-Hogg-Dubé symposium 2013 in Paris that the condition be called FLCN (folliculin) gene-associated syndrome (in short FLCN syndrome, or FLCN-S).
A definite diagnosis should ideally rely on the finding of a mutation in the FLCN gene and the presence of MCLD and/or multiple characteristic skin lesions and/or malignant kidney tumour. However although some diagnostic criteria have been proposed [17, 18] no consensus between geneticists, dermatologists, pulmonologists, and oncologists has hitherto been established.
Clinical Features
Pulmonary Manifestations
Pneumothorax is a characteristic feature [19–29] which has been reported as early as at age 7 years in the son of a patient [30]. In contrast patients may not develop pneumothorax throughout their lifetime as in a 85-year-old man with characteristic skin features present since young adulthood who had multiple pulmonary cysts detected by HRCT only once the diagnosis of FLCN-S was established in his son [31].
In a large series [22] of 98 patients affected with the FLCN-S, 13 carriers of FLCN haplotype, and 112 unaffected family members, the age-adjusted odds-ratio for pneumothorax in FLCN-S individuals was 50.3 times higher. In a larger cohort of 198 patients from the same institution, all 48 patients with a history of pneumothorax had multiple lung cysts identified by chest CT imaging [23]. The median age of occurrence of the first pneumothorax was 38 years (with a range from 22 to 71 years). Seventy five percent of patients had a second pneumothorax. There was no association between smoking and the risk of pneumothorax. Exon location of the BHD mutation was associated with the number of cysts, with individuals with mutations in exon 9 having more cysts. In a further study [24] of 51 families with FLCN-S, 88 % of the families and 84 % of the patients had lung cysts on CT imaging. Fifty-three percent of families and 38 % of individuals had a history of spontaneous pneumothorax. Most patients with a history of pneumothorax had lung cysts by CT imaging. Fifty-eight percent of patients with a family history of pneumothorax developed pneumothorax compared to 28 % without a family history (p = 0.01). A family history of pneumothorax was not associated with an increased risk of kidney tumours. Out of 34 individuals with a total of 92 spontaneous pneumothoraces, 19 had 2 pneumothoraces, and 7 had 3–7 pneumothoraces.
Interestingly, pneumothoraces without cysts on CT imaging have been reported [19]. A pneumothorax was reported in a 33 year-old-woman after a long airplane flight, leading to the diagnosis of MCLD and further FLCN-S [32]. Pneumomediastinum developed in a patient with FLCN mutation, without skin or renal manifestations, with pneumothoraces with or without detected lung cysts in four of family members [33].
Imaging
In a series of 17 patients with FLCN-S established by genetic testing [34], MCLD was present in 15 cases (bilateral in 13, and unilateral in 2). The distribution of the cysts predominated in the lower part of the lungs (13/15). Five patients had more than 20 cysts and 7 fewer than 10 cysts, the size of which ranged from 0.2 to 7.8 cm. The shape of the cysts varied from round to oval with the large cysts usually having a lobulated multiseptal appearance. In another study of 12 patients [35] the number of cysts varied from 29 to 407, with 77 % of cysts being of irregular shape, and 40 % located along the pleura. Distribution of cysts predominated in the lower medial zone (i.e. below the level of the tracheal carina, and in the inner half of the lung field). Cysts abutting or including the proximal portions of lower arteries or veins were present in all patients. The same group further compared the pulmonary cysts in patients with FLCN-S and in patients with LAM [36]. Females with FLCN-S were older (46 vs 36 year old), with a prevalence of 6/14 with a family history of pneumothorax within the second degree relatives (vs none in LAM patients). The percentage of the ratio of pulmonary cysts’ areas in each lung field, the number of cysts and the mean circularity of cysts (roundness) was greater in LAM; the mean size of cysts was greater in FLCN-S. The number and size of cysts in the reported studies is quite variable: some patients had only few cysts (5 or less to 10) whereas they were innumerable in other patients, with a size varying from a few millimetres up to 10 cm (Fig. 16.1).
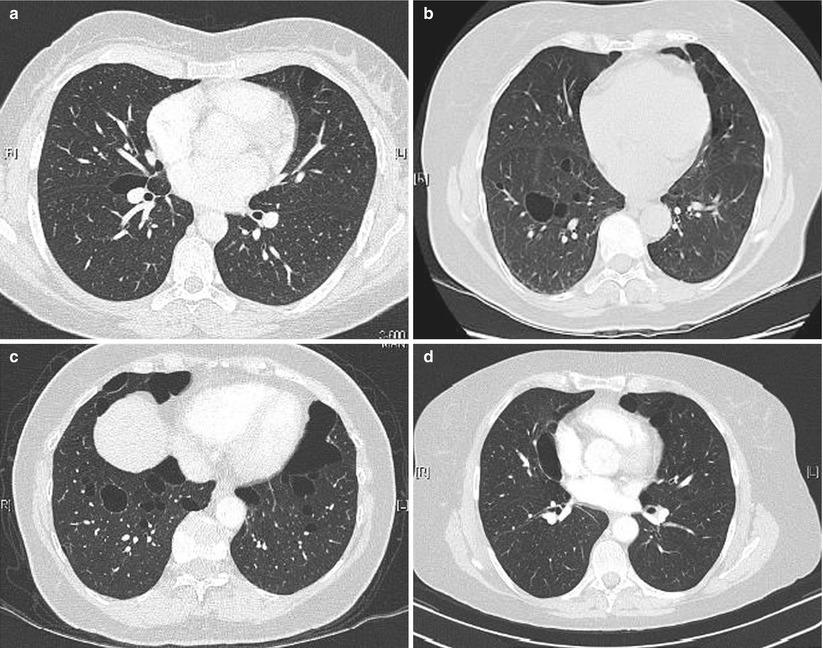
Fig. 16.1
Cysts in FLCN-syndrome. (a) Cyst of the lower medial zone, abutting the proximal portion of pulmonary vessels, oval shaped. (b) Cysts of the lower medial zone, round shaped. (c) Round, oval, and irregular shaped cysts. (d) Oval cyst bordering the pleura (left) and intraparenchymal cysts (which border the pleura)
Exon location of the FLCN mutation is associated with the number of cysts, with individuals with mutation in exon 9 having more cysts than individuals with mutation in other exons.
Pathology
Resected lung specimens of FLCN-S have often been diagnosed by pathologists as nonspecific blebs or bullae or emphysema [15]. The pathologic features mentioned in case reports usually did not identify specific lesions, however most often only specimen of subpleural lung tissue were taken at the time of pleurodesis after pneumothorax, with wedge lung biopsy further obtained in only a few cases. These reports mentioned possible associated inflammation attributed to chronic pneumothorax.
Histopathological study of two cases with basilar cysts found intraparenchymal collections of air surrounded by normal parenchyma or a thin fibrous wall, and blebs (consisting of collection of air within the pleura). Although these features were not specific to FLCN-S their predominantly basal location contrasted with the apical distribution of the other more common causes of spontaneous pneumothorax especially emphysematous bullae or idiopathic blebs [37]. The cysts in FLCN-S are typically punch out intraparenchymal cysts without inflammation [38]. A detailed analysis [39] was done on three wedge-resected tissues containing several cysts up to 1 cm in diameter, mostly in the pleural and subpleural regions. On microscopy these were found to be located in the vicinity of the interlobular septa, visceral pleura, and junctional regions between the interlobular septum and visceral pleura. Each cyst was incorporated with interstitial stroma of the interlobular septum and/or pleural in part, and with normal alveolar structures in the other part. Some cysts had protrusion of veins into the cystic space, a feature of an intimate spatial relationship of blood vessels and pulmonary cysts [28, 39, 40]. It was proposed that the pulmonary cysts in FLCN-S may represent an aberrant cystic alveolar formation with deranged interaction between alveolar epithelial structures and the surrounding mesenchyma in the peripheral lobular compartment that may give rise to the formation of abnormal cysts without stromal reaction. Pneumothorax might result from further growth of cysts [39]. Histopathological and morphometric analysis of 229 pulmonary cysts from 50 unrelated FLCN-S and 117 cysts from 34 patients with primary spontaneous pneumothorax were compared [41]. FLCN-S cysts differed significantly from primary spontaneous pneumothorax cysts by abutting on interlobular septa (88 %), having intracystic septa (14 %) or protruding venules (39 %) without cell proliferation or inflammation. Although the intrapulmonary FLCN-S cysts were smaller than the subpleural ones there was no difference in size between them when there was no inflammation. The conclusion of this study was that FLCN-S cysts are likely to develop in the periacinar region, an anatomically weak site in a primary lobule, where alveoli attach to connective tissue septa. The authors hypothesized that the FLCN-S cysts possibly expend in size as the alveolar walls disappear at the alveolar-septal junction, and grow even larger when several cysts merge. A study of eight cases reported that pulmonary cysts have their inner surface lined by epithelial cells, sometimes with a predominance of type II pneumocyte-like cuboidal cells with the cells constituting the cysts staining positive for phospho-S6 ribosomal protein expression, thus suggesting activation of the mTOR pathway [42]. Activation of the mTOR pathway has been considered in both TSC and FLCN-S. Facial hair follicle tumours (angiofibromas) in TSC patients coincidentally responded to the oral mTOR inhibitor rapamycin administrated after renal transplantation, and to topical rapamycin. However a topical rapamycin double-blind placebo-controlled randomised split-face trial did not demonstrate cosmetic improvement of fibrofolliculomas in FLCN-S patients [43]. Folliculin in the lung cysts studied by immunochemistry [39] was present in macrophages and epithelial cells in the lungs of the patients and normal control lungs. In the cyst lesions the pneumocytes that lined the inner surface of cysts were also immunostained for folliculin. Wedge biopsy in a patient with FLCN-S and a history of ten pack-years of smoking reported widespread emphysematous changes, with further an interlobular septum replaced by interstitial air resulting in the displacement and compression of the venous wall on the side abutting the air-filled space [25].
Cutaneous Manifestations
Characteristic multiple, dome-shaped, whitish papules usually develop after the age of 20 years, especially on the face (nose and cheeks). These are common on the neck, and they are sometimes present on the trunk or the ears. Histologically they consist of benign hair follicle tumours called fibrofolliculomas (Fig. 16.2).
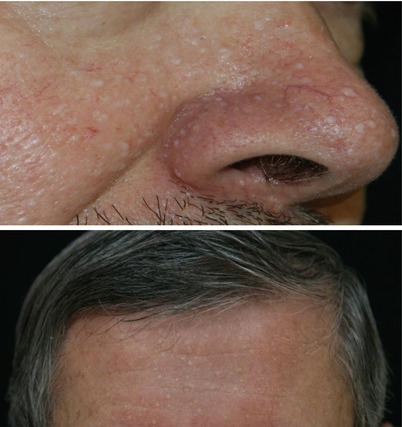
Fig. 16.2
Characteristic skin fibrofolliculomas in FLCN-S (Courtesy of D. Jullien, Lyon)
In their original description Birt et al. [6] described, in addition to fibrofolliculomas, trichodiscomas and acrochondromas. Whereas the first two belong to a morphological spectrum, acrochondromas are common in the general population. The diagnosis of the skin lesions relies on clinical examination by an experienced dermatologist with further histological examination if necessary.
Renal Manifestations
Renal cancer is the most threatening manifestation of the FLCN-S, occurring in up to one fourth of patients at a mean age of 50 years, and at a minimal age of 20. The most common histological types are chromophobe renal cancer, and a mixed pattern of chromophobe and oncocytic renal tumours. Renal cancer is multifocal and/or bilateral in more than half of patients with FLCN-S.
Most of these tumours are in the low-risk oncocytic renal neoplasia spectrum including chromophobe renal cell carcinoma, oncocytoma, and hydrid oncocytic tumours; clear cell and papillary renal cell carcinoma have also been reported [44, 45].
Renal (small size) angiomyolipoma [46] and renal cysts [12] has been reported in FLCN-S. Surveillance for renal tumours by annual magnetic resonance imaging starting by the age of 20 may be proposed.
Surveillance for renal tumours by annual magnetic resonance imaging starting by the age of 20 may be proposed.
Other Manifestations
Colorectal Polyps and Carcinoma
Colorectal polyps and carcinomas have been reported in patients with FLCN-S [9, 26, 48]. However systematic colonoscopy in 45 patients did not show an increased prevalence of colorectal neoplasm [22]. An increased risk of colorectal cancer might apply only to specific subgroups of patients with FLCN-S [26].
It has been suggested that FLCN is involved in the tumourigenesis of a subset of microsatellite stable sporadic colorectal carcinomas, and that the allelic loss in the region close to FLCN may play a role in colorectal tumour progression [49].
Clinical Vignette
A non-smoker woman, born 1951, had a first right pneumothorax in 1994 treated by intercostal tube drainage.
A relapse of pneumothorax (1995) led to thoracoscopic surgical resection of bullous dystrophy of the apical segment of the right upper lobe (no specific features present at pathological examination).
The patient further experienced 5 right pneumothoraces before she underwent in 2002 a right thoracotomy showing several subpleural bullous lesions. Pleurectomy was done.
She was referred for evaluation at the expert centre in 2011. HRCT demonstrated several pulmonary cysts with a maximum diameter of 18 mm. Lung function tests showed normal spirometry, moderate decrease of DLCO (81 % predicted), and no oxygen desaturation on exercise.
Dermatologic examination disclosed only two small papulous skin not characteristic of fibrofolliculoma lesions on the left cheek-bone (of which the patient refused biopsy).
No tumour was present at renal magnetic resonance imaging.
Heterozygous deletion of exon 14 of the FLCN gene led to the diagnosis of FLCN-syndrome (Birt-Hogg-Dubé).
FLCN Gene-Associated Familial Spontaneous Pneumothorax
A series of patients (24 females and 6 males) with FLCN mutations with pneumothorax and/or MCLD as the presenting feature was reported from the main institution in Japan where patients with suspected or diagnosed LAM are most often referred to [27]. Skin lesions of FLCN-S were present in only 7 and renal tumors in 2, and thus the majority of patients had neither skin nor renal involvement suggestive of FLCN-S. The same group further reported germline mutations of the FLCN gene in patients with multiple lung cysts and recurrent pneumothorax who had neither skin nor renal lesions [28].
A Chinese study reported patients with sporadic and familial isolated primary spontaneous pneumothorax with FLCN mutations [50]. Lung cysts were present but no other features were associated. FLCN deletion was reported in a large Finnish family with a dominantly inherited tendency to primary spontaneous pneumothorax [51]. All carriers of the deletion had between 1 and more than 30 cysts, 1–6 cm in diameter. The deletion was not present in unaffected family members or in control samples. As the Finnish family under investigation had only pulmonary manifestations, the absence of other FLCN-S symptoms could be due to the location of the deletion which occurred in the first coding exon (in contrast with almost all other mutations in FLCN which are truncations occurring downstream in the gene). A study of primary spontaneous pneumothorax in a Swiss family with FLCN mutation did not disclose other FLCN-S manifestations [52], as well as in another English family [53] where antenatal localised lobar cystic malformation was discovered in two patients (at 34 week gestation in one) which had no significant clinical consequences during life.
The above studies show that FLCN-S may manifest only with relapsing pneumothorax and MCLD, with occasionally minimal or no cutaneous manifestations.
Only limited information is available about the pulmonary functional consequences of FLCN -associated MCLD. With the exception of pneumothorax often relapsing, the pulmonary involvement is usually asymptomatic with lung function tests mostly normal or subnormal [25, 29]. No information on pulmonary prognosis is currently available.
Lymphangioleiomyomatosis
LAM is characterised by the proliferation of abnormal smooth muscle cells resulting in the formation of multiple lung cysts and further the possible development of chyle-filled cystic lesions in the axial lymphatics [54].
Sporadic LAM occurs almost exclusively in women, and in up to 40 % adult women with the genetic disease tuberous sclerosis complex. Sporadic and TSC-LAM are indistinguishable clinically and histologically, although in TSC-LAM multifocal micro nodular pneumocyte hyperplasia may also be present. Both TSC and LAM are associated with abnormalities in one of the two TSC genes (TSC1 and TSC2), in sporadic LAM generally TSC2 [55, 56].
Clinical Features
The first symptoms of LAM usually develop between 20 and 40 years of age however it may be recognised only after the menopause. Three-quarters of patients will experience pneumothorax and in about two-thirds of these it will be recurrent. Bilateral pneumothorax, pneumothorax during pregnancy, or recurrent pneumothorax in a young woman who does not smoke is suggestive of LAM. Most patients with LAM develop breathlessness generally of insidious onset. Occlusion of lymphatics by LAM cells can lead to chylous pleural effusions or chylous ascites. In patients with advanced pulmonary disease hypoxaemia may develop. Pulmonary hypertension secondary to hypoxaemia is usually mild.
Imaging
The pulmonary cysts in LAM have a size usually varying from 2 to 30 mm and are distributed diffusely and bilaterally throughout normal lung parenchyma. Their shape is mostly round (Fig. 16.3). In severe disease the parenchyma is replaced by cysts with a reticular “lace-like” aspect.
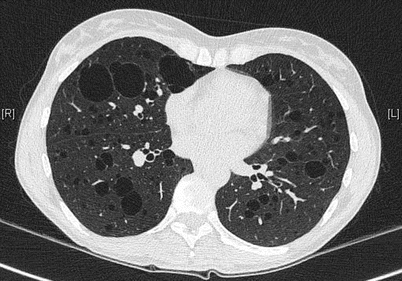
Fig. 16.3
Multiple cysts in LAM
Associated pneumothorax, or pleural effusion may be present.
In a series of 186 patients with TSC who had CT of the lung bases [57], thin wall cysts were found in 28 %. These were detected in 42 % of 95 females and also in 13 % of 91 males. Cysts were larger and more numerous in women than in men. Patients with TSC-LAM had, on average, slightly less severe lung disease than patients with sporadic LAM. Sixty-three percent of LAM patients had moderate to-severe disease compared with 40 % in TSC-LAM. Hepatic and renal AML and non calcified pulmonary nodules were more common in TSC-LAM, whereas lymphatic abnormalities (thoracic duct dilation, lymphangioleiomyomas) and chylous pleural and peritoneal effusion were more common in LAM.
Studies in a cohort of 293 patients from a TSC clinic provided more information on TSC-LAM according to the presence and type of mutations. Sixty-five patients with TSC were evaluated by chest CT [58] with cysts found in 49 % of women and 10 % of men. In the female population, changes consistent with LAM were present in 40 % with TSC1 mutations, 48 % with TSC2 mutation, and 71 % with no mutation identified. TSC2 women with LAM had a greater number of cysts than TSC1 women. Patients with TSC and no mutation have a higher incidence of both angiomyolipoma and LAM [59]. Among female patients with LAM, renal angiomyolipoma was universally present [60].
Multiple lung cysts may further be associated with multifocal micronodular lesions in TSC patients which consist of self-limited benign lesions with hyperplastic foci of large type II pneumocytes similar to atypical adenomatous hyperplasia of the lung, with a size up to 8–10 mm [61–63].
The imaging pattern of LAM at chest CT, although compatible and suggestive, is not pathognomonic and it can be mimicked by other lung conditions. The European Respiratory Society guidelines for the diagnosis and management of LAM recommended that a definite diagnosis be made in the presence of a characteristic CT scan (i.e. multiple >10) thin-walled round well-defined air-filled cysts with preserved or increased lung volume with no other significant pulmonary involvement especially no interstitial lung disease (with the exception of possible features of multifocal micronodular pneumocyte hyperplasia in patients with TSC) at CT scan, only when other supportive features of the disease are present namely, renal angiomyolipoma, TSC, a chylous collection or tissue biopsy from the lung or extra pulmonary sites consistent with LAM [64]. The combined prevalence of angiomyolipoma and lymphatic abnormalities mean that in over 50 % of cases an abdominal CT scan will be helpful in confirming the diagnosis of LAM without a lung biopsy [65]. A serum vascular endothelial growth factor-D (VEGF-D) level of greater than 800 pg/ml is found in approximately two-thirds of patients with LAM but not in other cystic lung diseases.
Patients with LAM generally have an accelerated loss of lung function with a rate of decline in forced expiratory volume in 1 s (FEV1) of 70–120 ml/year [66–68]. Some patients do not deteriorate over many years whilst others have a rapidly progressive course leading to respiratory failure within 5–10 years. Although at present there are no clear indicators of prognosis at diagnosis, presentation at a young age, an onset with breathlessness rather than pneumothorax, abnormal lung function at presentation including a low transfer coefficient for carbon monoxide (KCO) suggest more aggressive disease.
Pneumothorax is a particular problem for patients with LAM and early surgical intervention is appropriate. As the disease progresses patients develop worsening airflow obstruction, with 25 % of those partially responding to inhaled bronchodilators.
Overall, survival appears to be around 90 % at 10 years, with many patients having more longstanding disease [69].
Langerhans Cell Histiocytosis
Adult pulmonary Langerhans’ cell histiocytosis (PLCH), formerly called histiocytosis X, is a rare bronchiolar disorder developing almost exclusively in young smokers [70, 71].
Pathology
The Langerhans cell has a dendritic shape, a clefted nucleus, and immunohistochemical staining with anti-CD1a antibody which distinguishes it from other histiocytes. A further characteristic feature is the identification of intracellular Birbeck’s granules at electron microscopy.
The poorly demarcated granulomas formed by Langerhans cells extend from the bronchioles to the adjacent alveolar structures. The early cellular and granulomatous lesions are often centred on a cavity that is the residual lumen of the destroyed bronchiole. The end-stage lesions almost acellular consist of stellar fibrotic scars and/or cystic fibrous cavities [72].
Clinical Features
PLCH typically occurs in smokers 20–40 years old, with cough or progressive dyspnea on exertion, with pneumothorax leading to the diagnosis in about 10–20 % of patients. PLCH may also be diagnosed in a patient presenting with characteristic extra-pulmonary manifestations especially diabetes insipidus, cutaneous lesions, or osteolytic bone lesions. The lung function tests in PLCH may show airflow obstruction in patients with progressive disease. Reduction of carbon monoxide transfer factor is most common and may be severe. A discrepancy of subnormal FEV1/forced vital capacity (FVC) ratio with markedly decreased carbon monoxide transfer is common.
Imaging
On chest X-ray the pulmonary lesions predominate in the upper and middle lung fields (with sparing of the costophrenic areas). They consist of micronodular, reticular, or cystic features. CT imaging shows the presence of nodules (with a centrilobular distribution), cavitated nodules (thick-walled cysts), and thin-walled cysts. The nodules which are surrounded by normal parenchyma may be few or innumerable, with a size up to 20 mm. Cavitated nodules are very characteristic for PLCH. The thin-walled cysts often have bizarre irregular configurations with bilobed, cloverleaf, and branching morphologies.
Serial CT may typically show the characteristic sequence of nodules which cavitate into thick-walled cysts, and eventually thin-walled cysts. However the nodules may completely disappear without significant parenchymal sequelae.
In a series [73], the most common abnormalities consisted of micronodules ≤5 mm in diameter (89 %), thick-walled cysts (82 %), thin-walled cysts with walls of 2 mm or less (82 %), nodules (74 %), bizarre cysts (41 %), and reticulation (41 %).
Diagnosis and Management
The diagnosis of PLCH may confidently be established without lung biopsy when both cavitated nodules and thin-walled cysts are discovered on HRCT in young heavy smokers because of a pneumothorax or dyspnea, or when characteristic extra-thoracic features are present (isolated diabetes insipidus, or cutaneous involvement easily accessible to biopsy).
MCLD in Lymphoid Disorders
Lymphoid Interstitial Pneumonia
Lymphoid (lymphocytic) interstitial pneumonia results from diffuse reactive pulmonary lymphoid hyperplasia of bronchus-associated lymphoid tissue (BALT) with scattered plasma cells and histiocytes. Follicular bronchiolitis is a follicular hyperplasia of BALT present in the bronchiolar wall. LIP along with follicular bronchiolitis is considered as part of a spectrum of BALT hyperplasia related to connective tissue disease especially Sjögren syndrome [74] and other auto-immune disorders, and in infections with Epstein-Barr virus or human immunodeficiency virus.
Cysts were present in 15 patients in a series of 22 patients with LIP [75] including 6/10 with Sjögren syndrome (SS), 6/7 with Castleman disease, 1/2 with acquired immunodeficiency syndrome (AIDS), and 2/3 with no underlying disease. The average size of the cysts was 6 mm, these being bilateral in 10 and unilateral in 5. Other common findings in this series included areas of ground-glass attenuation and poorly defined centrilobular nodules in all the patients, subpleural small nodules, thickening of bronchovascular bundles, interlobular septal thickening, and lymph node enlargement.
In a large study [76] of 80 patients with Sjögren syndrome (56 primary including 11 with clonally derived lymphoproliferative disorder, and 24 secondary), cysts were present in 30 (22 in primary and 8 in secondary). Other CT findings consisted of interlobular septal thickening, intralobular reticular opacities, ground-glass opacities, honeycombing. The presence of cysts was independently and significantly associated with clonal lymphoproliferative disorder and anti-SSB antibody seropositivity. The mean number of cysts was 14, with a mean size of 16 mm, predominating in the lower and outer one-third zones.
Cysts in patients with primary SS were reported in 18/60 patients and were significantly associated with anti-SSB antibodies and clonally derived lymphoproliferative disorder [77]. Multiple bullae were present on CT imaging in only 3/32 patients with primary SS in another series [78]. Cysts in Sjögren syndrome are presented below (Fig. 16.4).
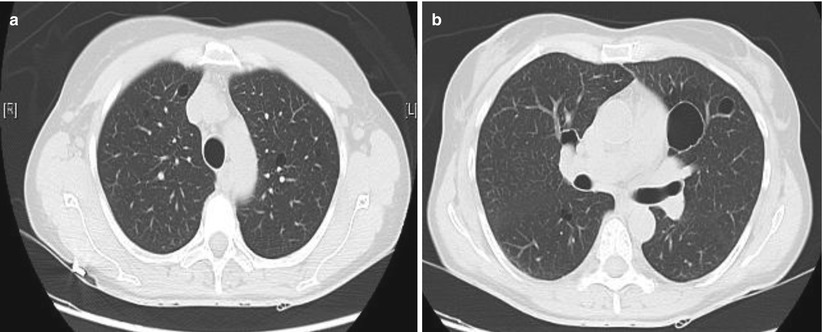
Fig. 16.4
Cysts in Sjögren syndrome. (a) Small intra-parenchymal round cysts. (b) More voluminous cyst in the same patient
Pulmonary Lymphoma
Low-grade lymphoma of the BALT is a rare cause of MCLD. The distribution of the lesions is centred on the airways with features of bronchiolitis with possible associated airflow obstruction [79, 80]. BALT lymphoma is diagnosed by immunohistochemistry staining monotypic B cell markers. However the definite proof of clonal proliferation is obtained only by polymerase chain reaction-based DNA testing for immunoglobulin chains and T-cell gene rearrangements. A monoclonal gammopathy when present in the blood and/or urine may orientate the diagnosis.
Amyloidosis and Nonamyloid Immunoglobulin Deposition Disease
Amyloidosis
MCLD in isolated pulmonary lymphoma may be associated with amyloidosis [81]. The cystic lesions were found to correspond to dilated bronchioles infiltrated by amyloidosis. Multiple cysts associated with nodular amyloidosis (of the AL type) especially in Sjögren syndrome without systemic amyloidosis [82–85], and isolated multiple cystic pulmonary amyloidosis have been reported [86].
Diffuse alveolar septal amyloidosis with multiple cysts and calcification has only rarely been reported [87].
NonAmyloid Immunoglobulin Deposition Disease
Whereas light-chains of monotypic immunoglobulins may deposit in the tissues as fibrillar amyloidosis resulting from a β-pleated sheet configuration consequently binding Congo red, less commonly, light-chains may deposit in the tissues (especially the kidney) as an amorphous (non fibrillar) material [88].
Light chain deposition has been described in patients with MCLD referred for lung transplantation with a presumptive diagnosis of either PLCH or LAM. The disease was histologically characterised by non-amyloid deposits in the alveolar walls, the small-airways and the vessels (with further emphysematous-like changes and small airway dilation). Electron microscopy revealed coarsely granular deposits. Monotypic kappa light-chain fixation was demonstrated on the abnormal deposits and along the basement membranes [89]. Further using polymerase chain reaction a dominant lymphoid B-cell clone was identified in the lung with biological features suggesting antigen-driven primary pulmonary lymphoproliferative disorder [90]. A monoclonal immunoglobulin component may be identified in the blood and/or urine in a proportion of patients. The diagnosis of light-chain deposition disease may be obtained by pulmonary biopsy but also by bronchial biopsy in non-amyloid immunoglobulin deposition disease presenting as MCLD [91]. Mass spectrometry on formalin fixed paraffin-embedded tissue may confidently diagnose kappa light chain deposition disease [92]. Cysts in non-amyloid monoclonal immunoglobulin deposition disease are presented above (Fig. 16.5)
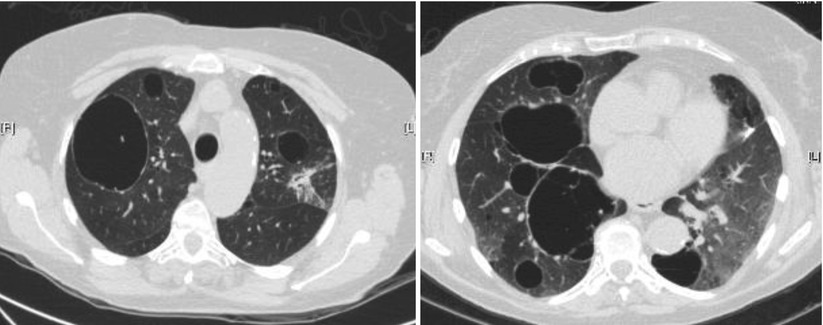
Fig. 16.5
Cysts in non-amyloid monoclonal immunoglobulin deposition disease (same patient)
MCLD of Infectious Origin
Cysts of infectious origin are often referred to as pneumatoceles which result from an inflammatory process that causes central parenchymal necrosis, with a cystic expansion due to elastic retraction of the surrounding lung tissue. These often occur in patients in whom only a small area of bacterial pneumonia with consolidation was present. The cyst is frequently larger than the preceding infiltrate and characteriscally has thin walls.
Staphylococcal and Other Bacterial MCLD
Pneumatoceles develop rapidly and often resolve spontaneously [93].
Pneumonia due to Staphylococcus aureus especially common in the middle of the last century was characteristically associated with multiple pneumatoceles. The mere presence of pneumatoceles on chest X-ray was then commonly considered as diagnostic of staphylococcal pneumonia. They usually develop in the first week of pneumonia and disappear in an average of 6 weeks [94–97].
Pneumatoceles have also been reported in other bacterial pneumonias, especially in pneumococcal pneumonia in up to 19 % of cases [98].
Pneumocystis jiroveci Associated MCLD
Pneumocystis jiroveci pneumonia, occurring in immunocompromised patients, was a major cause of morbidity and mortality in patients with acquired immunodeficiency syndrome (AIDS) in the 1980s [99]. Since antiretroviral therapies have been available, the incidence of P. jiroveci pulmonary infection and mortality in AIDS in developed countries have dramatically decreased.
In AIDS patients the typical pulmonary features of P. jiroveci pulmonary infection consist of diffuse pulmonary ground-glass and/or reticular opacities usually without pleural effusion. Pulmonary cysts, thin or thick walled, with regular or irregular shape, may be present either isolated or more commonly associated with infiltrative opacities. Cysts may regress under highly active antiretroviral therapy [100].
In a comparative study [101] of P. jiroveci in 38 immunocompromised patients, those with AIDS had a higher proportion of cysts (56 vs 3 %, p = 0.015) and a lower proportion of ground glass attenuation 44 vs 86 % (p = 0.02). On multivariate analysis, only AIDS was a risk factor for the formation of cysts.
Recurrent Respiratory Papillomatosis
Recurrent respiratory papillomatosis (RRP) caused by human papilloma virus (HPV) especially types 6 and 11 develops in the upper airways especially the larynx. It usually presents in childhood and is thought to be acquired through vaginal birth, especially from mothers with condyloma. Tracheal and bronchopulmonary spread of RRP developed in only 1.8 % of a series of 448 patients with juvenile onset RRP [102]. The pulmonary lesions characteristically consist of bilateral multiple cavities, thin-walled cysts, and nodules predominating in the lower lobes [103–105]. The cysts are usually less than 5 cm in diameter, with occasional air-fluid level (resulting from superimposed infection). The destruction of the lungs by papillomatosis may occasionally be massive. Mortality from pulmonary papillomatosis is high resulting from the extensive cystic destruction of the lungs. Malignant transformation into squamous cell carcinoma may occur [106].
MCLD in Hyper-IgE Syndrome
The hyper-IgE syndrome is a primary immunodeficiency characterised by very high levels of Serum IgE (mean about 20,000 UI/mL), eczema, recurrent infections (especially pulmonary and cutaneous), with further connective tissue and skeletal abnormalities. Recurrent skin abscesses and pneumonia result mainly from Staphylococcus aureus infection. Mutations in the gene encoding the signal transducer and activator of transcription 3 in the type 1 of the syndrome (classic autosomal dominant or sporadic) have been reported [107].
Pneumonia onset ranges especially from the newborn period to 3 years, with the formation of cysts (solitary or multiple) up to 8 years after pneumonia. The size of the cysts may be up to 12 cm. They may resolve after prolonged antibiotic therapy. The histopathologic features of the cysts are those of chronic abscesses with a dense necrotic layer of exudates with leukocytic infiltration [108–110].
MCLD of Tumoural Origin
MCLD has been reported especially in patients with sarcoma and in patients with benign uterine leiomyoma.
Metastases of Sarcomas
Pneumothorax may be the presenting manifestation of an extrapulmonary sarcoma with cystic metastases [111, 112]. Pneumothoraces in patients with pulmonary cystic metastases from sarcoma have been reported in patients with angiosarcoma [113–116], epithelioid sarcoma [117–120], leiomyosarcoma [112], osteosarcoma [121]. The wall of the cysts is usually composed of tumor cells.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


