1. Age at disease onset <40 years
Development of symptoms or findings related to Takayasu arteritis at age <40 years
2. Claudication of extremities
Development and worsening of fatigue and discomfort in muscles of 1 or more extremity while in use, especially the upper extremities
3. Decreased brachial artery pulse
Decreased pulsation of 1 or both brachial arteries
4. BP difference >10 mmHg
Difference of >10 mmHg in systolic blood pressure between arms
5. Bruit over subclavian arteries or aorta
Bruit audible on auscultation over 1 or both subclavian arteries or abdominal aorta
6. Arteriogram abnormality
Arteriographic narrowing or occlusion of the entire aorta, its primary branches, or large arteries in the proximal upper or low extremities, not due to arteriosclerosis, fibromuscular dysplasia, or similar causes; changes usually focal or segmental
For purposes of classification, a patient shall be said to have Takayasu arteritis if at least 3 of these 6 criteria are present. The presence of any 3 or more criteria yields a sensitivity of 90.5 % and a specificity of 97.8 %
Epidemiology
Although TA has a worldwide distribution, the disease is thought to be more prevalent in Asian, Middle-East, and Central and South American countries than in North America [6–8] or Europe [3], with some differences in the characteristics of the disease among the various ethnic backgrounds [3, 7, 9, 10]. The greatest frequency of the disease is observed in Japan [11, 12]. Conversely, the incidence of the disease is as low as 0.3–2.6 cases per million per year in the USA, Sweden, Germany and UK [3], suggesting that TA is one of the most infrequent form of vasculitis. One of the typical epidemiological features of TA is the marked predominance of the disease in women, with a F/M sex-ratio known to vary from 29/1 to 1.2/1 [3]. This very wide range may reflect either biases in case collection or differences between ethnic groups. Most TA patients have disease onset during the second or third decade of life. However, neither the occurrence of TA in patients over 50 years nor in children is uncommon [3, 13].
Pathologic Features
Because biopsy of involved vessels is not usually performed in TA, the diagnosis mostly relies on clinical features and vascular imaging. Pathologic findings in the pulmonary arteries have been poorly documented [14] and most available data originate from other arteries. Active inflammation in TA is typically indicated by the presence of mononuclear cells within the vascular wall, predominantly lymphocytes and macrophages. These cells are mostly recruited in the media and adventitia through the vasa vasorum. Because TA is a granulomatous vasculitis, giant cells and granulomas are commonly found in the media during active inflammation. Intimal proliferation contributes to the development of stenotic arterial lesions. At a more advanced stage, pathologic features include vascular wall fibrosis, while the destruction of the elastic lamina and the muscular media can lead to aneurismal dilation of the affected vessel. Retrospectively, dense scar tissue remains as an indication of prior vasculitis.
Pathogenesis
While our knowledge of the pathogenesis of TA has considerably improved during the last decade, the exact pathogenic sequence and natural history of vascular lesions remain unknown. By using cluster analysis we have recently shown that paired vascular beds usually clustered with their contralateral counterparts, while vascular lesions extended contiguously in the aorta [15]. Cell-mediated mechanisms are thought to be of primary importance in TA (Fig. 11.1). Therefore, it is currently hypothesized [15] that an unknown stimulus triggers the expression of the 65 kDa Heat-shock protein in the aortic tissue which, in turn, induces the Major Histocompatibility Class I Chain-Related A (MICA) on vascular cells. The γδ T cells and NK cells expressing the NKG2D receptors recognize MICA on vascular smooth muscle cells and release perforin, resulting in acute vascular inflammation. Pro-inflammatory cytokines and chemokines are therefore released and increase the recruitment of mononuclear cells within the vascular wall. Then, T cells infiltrate and recognize one or a few antigens that could be presented by a shared epitope, which is associated with specific major Histocompatibility Complex alleles on the dendritic cells, these latter being activated through their Toll-like receptors. Th1 lymphocytes drive the formation of giant cells through the production of interferon-γ, and activate macrophages with release of vascular endothelial growth factor (VEGF) resulting in increased revascularization and platelet derived growth factor (PDGF), resulting in smooth muscle migration and intimal proliferation. Th17 cells induced by the IL-23 microenvironment may also contribute to vascular lesions through activation of infiltrating neutrophils. Although being very controversial, dendritic cells may cooperate with B lymphocytes and trigger the production of anti-endothelial cell auto-antibodies resulting in complement-dependent cytotoxicity against endothelial cells.
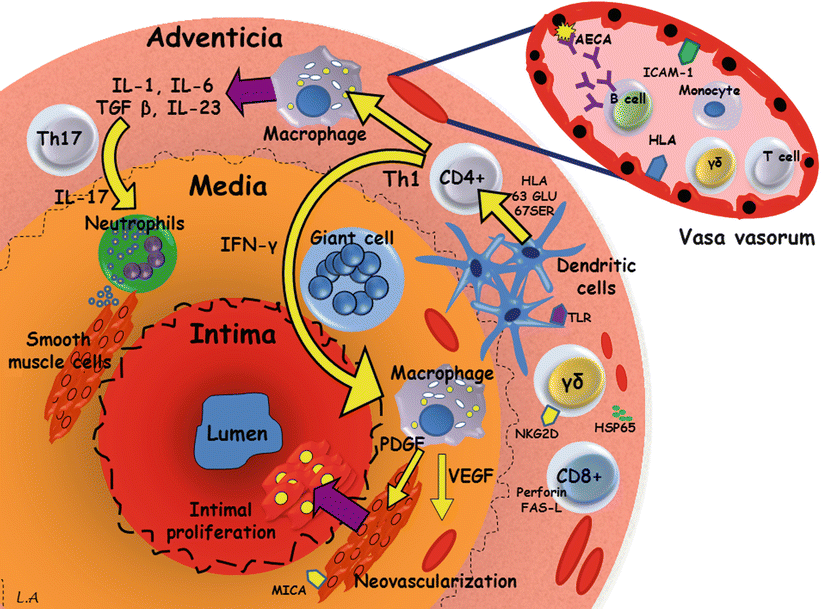
Fig. 11.1
Pathogenesis of Takayasu’s arteritis. AECA anti-endothelial cell antibodies, FAS-L FAS ligand, HLA human leukocyte antigen, HSP65 heat-shock protein 65, ICAM-1 intercellular adhesion molecule 1, IFN interferon, IL interleukin, TLR toll-like receptors, MICA major histocompatibility complex class I-related chain A, NKG2D natural killer group 2, member D, PDGF platelet-derived growth factor, TGF transforming growth factor, VEGF vascular endothelial growth factor, γδ gamma-delta cell
Clinical Vignette
A 23-year female originating from Madagascar was referred for fatigue, hypertension, lower limb claudication, and long-standing low-grade fever. Clinical examination revealed diffuse vascular bruits over the carotid arteries, abdominal aorta and iliac arteries, blood pressure asymmetry over 10 mmHg and diminished popliteal, posterior tibial and dorsalis pedis pulses. Laboratory examination revealed raised acute phase reactants (ESR: 60 mm/1st h, CRP: 5 mg/dL). Computed Tomography angiography showed typically thickened thoracic and abdominal aortic wall with subocclusive stenoses of the iliac arteries. Echocardiography was normal. Extensive workup ruled out any ongoing infectious disease. Diagnosis of Takayasu’s arteritis was made and she was treated with prednisone 1 mg/kg/day orally followed by slow tapering and tuberculosis prophylaxis (because she was originating from an area where tuberculosis is highly prevalent). Her condition markedly improved within 3 weeks and follow-up at 6 months revealed significant improvement of arterial lesions. Unfortunately, lower limb claudication reoccurred when corticosteroids were tapered down to 15 mg/day. Therefore, prednisone was increased back to 30 mg/kg/day and azathioprine 3 mg/kg/day was added. Corticosteroids were slowly tapered again and azathioprine eventually stopped. Three years later, she is totally asymptomatic under prednisone 5 mg/kg/day, which is our consolidation regimen.
Clinical Features
The clinical course of TA is classically thought to progress through three distinct stages: first, an early phase with prominent constitutional and systemic symptoms such as fatigue, weight loss, fever, and arthralgia; second a vascular phase occurring months or years later, with clinical manifestations of ischemia due to stenotic or occlusive lesions, or related to aneurysms; and third, a late phase (also called “burnt out phase”) with fibrotic and fixed vascular abnormalities [6]. While more than 90 % of patients have vascular signs or symptoms during the course of the disease, it is now well recognized that the systemic and vascular phases may overlap, and that a significant proportion of patients may never exhibit any constitutional symptom [3, 6].
The clinical presentation of TA is heterogeneous, and comprises many non-specific findings such as constitutional symptoms (fatigue, fever and weight loss), musculoskeletal features (arthralgia, arthritis), cardiac and vascular features (vascular bruit, blood pressure asymmetry, claudication of extremities, carotodynia, hypertension, valvular involvement with aortic regurgitation, Raynaud’s phenomenon, pericarditis), neurologic features (headache, visual disturbance, stroke or transient ischemic attacks, seizures), dermatologic manifestations (erythema nodosum, pyoderma gangrenosum).
Pulmonary artery involvement of TA is believed to occur in 15–65 % of patients [11, 14, 16–22]. It may occasionally be the revealing [23, 24] or foreground feature of the disease [23, 25, 26]. Clinical signs of pulmonary involvement in TA are usually non-specific and therefore may lead to delayed diagnosis [3, 27]. These mostly include chest pain, cough, signs of pulmonary hypertension such as dyspnea, fatigue, angina, syncope [28–31], and hemoptysis [32, 33], with rare cases of pulmonary hemorrhage [34–36]. The exact frequency of pulmonary hypertension is unknown in TA, and is mostly due to pulmonary stenosis or left heart involvement [37]. However, other causes, including pulmonary capillary haemangiomatosis have been occasionally reported [38]. In a study of 76 Mexican TA patients [39], 10 (13 %) developed pulmonary hypertension using transthoracic echocardiography. Pulmonary artery hypertension was observed in 20 % of patients with pulmonary artery involvement among patients with pulmonary artery involvement reported in a Chinese study published in 1994 [19]. Pulmonary hypertension in TA was statistically associated with disease activity in a Korean series of 204 patients [40], those with active disease (defined as patients having an elevated ESR or CRP level, thickened arterial wall with mural enhancement on CT or MR angiography, and carotidynia at the time of the initial diagnosis) had a higher incidence of pulmonary hypertension than those with inactive disease. In a Japanese study [41], a significant correlation was found between plasma endothelin-1 levels, which is involved in the pathogenesis of pulmonary hypertension, and erythrocyte sedimentation rates. Occasionally, clinical and radiographic features mimicking pulmonary embolism may be the first manifestation of Takayasu’s arteritis [42–44], and pulmonary infarction may occur in this setting [45–47]. Rarely, coronary artery to pulmonary artery collaterals may develop and induce coronary steal and myocardial ischemia [48, 49].
Laboratory Findings
Dealing with TA patients is challenging because there is no sensitive or specific biologic markers for diagnosis and monitoring disease activity in TA [50]. It is well known that clinical assessment alone may underestimate disease activity [6, 51] and current disease activity criteria (Table 11.2) are non-validated [6]. Previous studies have shown that ESR and CRP did not correlate with clinical features in about 50 % of cases [6, 7]. Interleukin-6, RANTES (Regulated upon Activation, Normal T Cell Expressed and Secreted), and Pentraxin-3 blood levels are believed to correlate with disease activity, but these markers are not widely available [52, 53].
Systemic features, such as fever, musculoskeletal (no other cause identified) |
Elevated erythrocyte sedimentation rate |
Features of vascular ischemia or inflammation such as claudication, diminished or abolished pulse, bruit, vascular pain (carotodynia), asymmetric blood pressure in either upper or lower limbs (or both) |
Typical angiographic features |
Imaging Studies
Because the clinical presentation and results of laboratory tests are typically nonspecific, accurate diagnosis of TA commonly depends on imaging studies. While conventional angiography has for long been the “gold standard”, this imaging modality is now outdated, while Computed Tomography (CT) and Magnetic Resonance Imaging (MRI) angiographies are increasingly used (Figs. 11.2, 11.3 and 11.4). These latter offer several advantages, including their non-invasiveness and their capability to demonstrate both mural and luminal changes in the pulmonary arteries, which is of major interest in TA because luminal changes may be delayed [22, 23, 54, 55]. Recently, pulmonary perfusion MRI has been shown to be a new alternative for the evaluation of pulmonary perfusion in TA [22, 56]. In a Japanese study [56], pulmonary MR perfusion images were acquired in 21 TA patients. The presence of perfusion abnormality was determined in both lobe-based (n = 126) and patient-based (n = 21) analyses. Sensitivity, specificity, positive predictive value (PPV), and negative predictive values (NPV) were calculated using perfusion scintigraphy as a standard reference. For lobe-based analysis, sensitivity was 91.7–95.8 %, specificity was 92.2–93.7 %, and PPV and NPV were 73.3–76.7 % and 97.9–99.0 %, respectively. For patient-based analyses, sensitivity was 100 %, specificity was 72.7 %, and PPV and NPV were 76.9 and 100 %, respectively. Therefore MR perfusion imaging appeared to be a valuable, non-invasive method to estimate pulmonary artery involvement in TA patients. Pulmonary perfusion scintigraphy has been shown effective to detect presence of pulmonary artery involvement in TA [13, 17, 18, 20, 57] and correlates well with pulmonary angiography [58]. Another convenient way to assess vascular involvement in TA is peripheral vascular Doppler. Unfortunately, vascular Doppler is only suitable for following peripheral disease progression and not pulmonary involvement in TA. 18F-Fluorodeoxyglucose-positron emission tomography (FDG-PET) scanning has been proposed as a new way of assessing disease activity in TA, but its role in the diagnostic and follow-up strategy remains controversial [59].
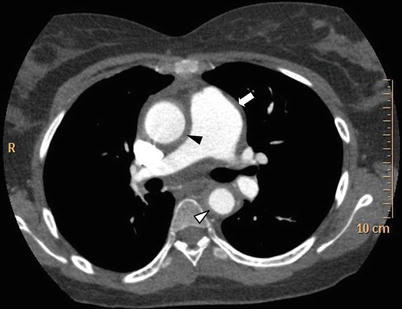
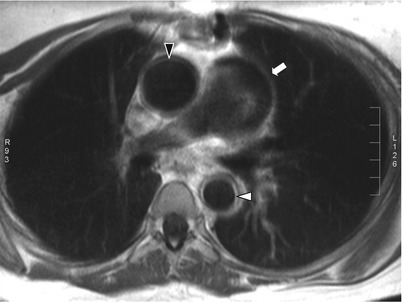
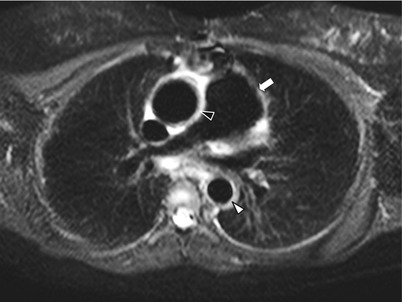
Fig. 11.2
Thoracic CT scan in Takayasu arteritis. Thoracic CT scan in mediastinal windows in an 18-year old woman with TA showing dilatation of the ascending aorta and increased thickening of the ascending aorta wall (black arrowhead), of the thoracic descending aorta wall (white arrowhead) as well as thickening and dilatation of pulmonary trunk (white arrow)
Fig. 11.3
Contrast-enhanced thoracic MRI in Takayasu arteritis. Contrast-enhanced T1-weighted black-blood thoracic MRI (axial view) in an 18-year old woman with TA showing dilatation of the ascending aorta and increased thickening of the ascending aorta wall with ring-like contrast enhancement (black arrowhead), and increased thickening of the thoracic descending aorta wall with ring-like contrast enhancement (white arrowhead) as well as thickening and dilatation of pulmonary trunk (white arrow)
Fig. 11.4
STIR thoracic MRI in Takayasu arteritis. Short tau inversion recovery (STIR) thoracic MRI (axial view) in an 18-year old woman with TA showing dilatation of the ascending aorta and increased thickening of the ascending aorta wall with hyperintense signal (black arrowhead), and increased thickening of the thoracic descending aorta wall with hyperintense signal (white arrowhead) as well as thickening and dilatation of pulmonary trunk (white arrow)
The most characteristic imaging findings of pulmonary involvement in TA include wall thickening and enhancement in early phases, stenotic or occlusive changes during the vascular phase, and fibrotic and/or calcified stenosis or occlusion in the chronic phase. These lesions mainly involve the segmental and subsegmental arteries, and less commonly the lobar or main pulmonary arteries [60–62]. Unilateral occlusion of a pulmonary artery can occur in advanced cases. Therefore, late-phase TA should always be considered in cases of chronic pulmonary artery obstruction of unknown origin [60]. Occasionally, TA may mimic unilateral pulmonary artery agenesis in children [63]. While being frequent in the aorta and its main branches, vascular dilatation and aneurysms in the pulmonary arteries are uncommon findings among TA patients [14, 16, 19, 21]. In a Chinese study performed in 1994 [19], pulmonary artery involvement was assessed by conventional digital subtraction angiography arteriography in 45 (33.8 %) of 133 patients. Stenosis and/or occlusion of segmental and/or lobar pulmonary arteries, and subsegmental branches, were the basic angiographic findings. Pulmonary artery branches in the upper lobes were more commonly affected than those in the lower and middle lobes. Bilateral lesions were more common than unilateral ones. Single lobar and segmental lesions were rare. No main pulmonary artery involvement was detected. In a Japanese study published in 1992 [14], 21 of 30 patients (70 %) had pulmonary artery involvement at pulmonary arteriography. Abnormalities were most common in upper lobe pulmonary arterial branches and segmental branches, followed by subsegmental branches. Systemic artery-pulmonary artery communications were seen in 6 patients (20 %). While regional hypoperfusion due to pulmonary arteritis may occasionally be seen on lung CT-scan [64], involvement of the lung parenchyma is atypical in TA, and alternative diagnoses should also be considered [65–67].
Therapeutic Management
Corticosteroids are usually considered the mainstay of medical treatment in active TA with early phase or vascular phase manifestations [68, 69]. However, approximately 40–50 % of patients require additional immunosuppressive agents to achieve and maintain remission [3, 7, 70]. Corticosteroids and other immunosuppressive agents have been shown effective to treat pulmonary involvement of TA, as these regimen may reverse pulmonary artery stenosis [28, 71]. Azathioprine [72], methotrexate [73], mycophenolate mofetil [74, 75], cyclophosphamide [76], and anti-TNFα agents [20, 77, 78] have all been recognized effective in open-label trials but no randomized controlled trial data are available. Therefore, the therapeutic choice should be mainly guided by the individual benefit-risk ratio. There is no specific recommendations for the treatment of TA-associated pulmonary hypertension, therefore the latter should be treated according to the current standard of care, while it may occasionally be intractable [29].
Treatment with corticosteroids and immunosuppressive agents is not mandatory during the late phase, as only limited improvement, if any, can be achieved for fibrotic and fixed vascular abnormalities. However, both angioplasty and stenting [28, 79, 80] or vascular bypass [31, 81] may be necessary in TA patients when symptomatic and/or hemodynamically significant stenoses, including pulmonary arterial stenoses, have occurred. Pulmonary artery bypass has been shown effective for in-stent stenosis following angioplasty for isolated pulmonary TA [82]. Bronchial artery embolization may be considered in case of intractable hemoptysis [83].
Prognosis
The early, intermediate and long term outcome of angioplasty and vascular bypass procedures in TA is generally considered satisfactory [51, 71, 82, 84–95], even for treatment of pulmonary artery involvement where available experience is more limited [80].
Comparison of survival between TA series is likely biased because different enrollment criteria and care strategies have been used. In most series from developed countries, the 5 and 10-year survival rates are of ≈95 and ≈90 %, respectively [3, 6–8, 96–98]. Park et al. [96] underlined that major prognostic factors in TA were presence of valvular heart disease, cerebrovascular accidents, congestive heart failure, ischemic heat disease, retinopathy and renovascular hypertension [96]. However, the exact prognostic value of pulmonary involvement in TA remains currently unknown.
Behçet’s Disease
Behçet’s disease (BD) is a multisystem and chronic disease of unknown etiology characterized by relapsing manifestations, including oral and genital ulcers, uveitis, and vasculitis, cutaneous, articular and central nervous system involvement.
BD’s vasculitis is particularly distinctive because both veins and arteries can be affected, mainly in the form of arterial aneurysms and of venous or arterial thrombosis. Pulmonary involvement in BD can almost be summarized as pulmonary artery aneurysm and pulmonary thrombosis. Parenchymal manifestations are less documented and can be either isolated or associated with pulmonary arterial involvement, therefore being a direct consequence of parenchymal ischemia.
Epidemiology
BD occurs worldwide but is most prevalent in the countries of the ancient Silk Road, especially in Turkey with 20–240 cases per 100,000 [99]. Prevalence ranges from 13.5 to 22 cases per 100,000 in Middle East and Asian countries. It is less frequent in Western countries, preferentially affecting migrants from endemic countries with a prevalence ranging from 0.12 to 0.64 per 100,000 [99].
The disease typically affects young adults from 20 to 50 years. BD is as frequent in male as in female but more severe in the former, with increased mortality, and more frequent ocular, major vessel or neurologic involvement [100].
Pathologic Features
Characteristic histopathological features of BD are vasculitis and perivascular inflammatory infiltrates of neutrophils and T-cells [101]. Vasculitis can involve both veins and arteries of all sizes [102]. Studies of pulmonary artery aneurysms reveal perivascular infiltrates and small-vessel vasculitis in the vasa vasorum [103], marked intimal thickening with disruption of the elastic lamina, degeneration of the tunica media, and thrombotic occlusion with recanalization [104].
Pathogenesis
While the pathogenesis of BD remains largely unknown, the disease is thought to be at the frontline between autoimmune and auto-inflammatory diseases [105]. Like many chronic inflammatory diseases, BD is believed to be the consequence of interplay between genetic susceptibility and environmental factors (mainly bacterial infections) [106]. There is a close association between HLA-B51/B5 and BD, suggesting a pivotal role of these alleles in the pathogenesis of the disease [107]. Many infectious agents have been implicated in the pathogenesis of BD [99], with Herpes simplex virus and Streptoccocus sanguis being most consistent candidates. Both are mainly found in oral mucosa and thus could explain the prominent feature of oral ulcers [108]. However, none of these microorganisms has proven to be the causative agent of BD and it has been hypothesized that many antigens, including bacteria heat-shock proteins, could trigger immune cross-reactive responses [109].
Main pathogenic features of BD are vasculitis, neutrophils hyperactivity and aberrant immunological responses [99]. Perivasculitis is found in BD lesions, including oral and genital ulcers, posterior uveitis and neurologic lesions [110]. Tissue injury seems to be the result of neutrophils infiltration and overproduction of superoxide and lysosomal enzymes [111]. High levels of pro-inflammatory cytokines (TNF, IL-1β, IL-8) have been measured in patients’ serum and could explain enhanced chemotaxis [99]. The recruitment of neutrophils within affected tissues could be under control of IL-17 producing T-cells [112, 113]. Pivotal role of gamma-delta T cells have also been shown. Activation of this innate population of T cells by microbial antigens could be the missing link between infectious agents and overreacting neutrophils [114].
Diagnostic Criteria
BD should be considered a diagnosis of exclusion, without any available pathognomonic diagnostic test. International diagnostic criteria were adopted in 1990 (International Study Group, ISG criteria, Table 11.3) [115]. In addition to oral ulcerations, which are a prerequisite, diagnosis of BD requires two of the following features: genital ulcerations, eye lesions, positive pathergy test or skin lesions (folliculitis or erythema nodosum). Pathergy test is not commonly used in Western countries because of its frequent negativity [116]. Altogether, these criteria are questionable as they sometimes fail to diagnose cases of BD without prominent mucocutaneous manifestations, for example cases where foreground feature is vascular involvement [117].
Table 11.3
International diagnosis criteria of Behçet’s disease, International Study Group for Behçet’s Disease
Recurrent oral ulceration | Minor aphthous, major aphthous, or herpetiform ulceration observed by physician or patient, which recurred at least 3 times in one 12-month period |
Plus 2 of the following, in absence of other clinical explication | |
Recurrent genital ulceration | Aphthous ulceration or scarring, observed by physician or patient |
Eye lesions | Anterior uveitis or posterior uveitis, or cells in vitreous on slit lamp examination or retinal vasculitis observed by ophthalmologist |
Skin lesions | Erythema nodosum observed by physician or patient, pseudofolliculitis or papulopustular lesions or acneiform nodules observed by physician in postadolescent patients not on corticosteroid treatment |
Positive pathergy test | Read by physician at 24–48 h |
Clinical Features
In the absence of any pathognomonic laboratory test, diagnosis of BD is strictly clinical and requires careful bedside evaluation [99]. In case of inaugural pulmonary manifestations, the diagnosis mostly relies on extra-pulmonary features, as the former are mostly non-specific. It is therefore important to look for a history of recurrent oral and genital ulcers, episodes of eye inflammation or visual loss, and past history of venous or arterial thrombosis. Genital scares are the hallmark of previous BD’s flares and must be carefully searched for. Skin examination must look for erythema nodosum and pseudo-folliculitis. Although pathergy test is not often performed in Western countries, hypersensitivity at puncture point can be found in the form of a small pustule. Eye examination is mandatory. Previous uveitis flares can present as anterior synechia. Moreover, retinal vasculitis can be asymptomatic but threatens the visual prognosis.
Prevalence of pulmonary involvement in BD seems to range from 1 to 18 % [118]. It mostly affects young males like other severe manifestations of BD [119].
Although inconstant and poorly specific, haemoptysis is the most frequent revealing symptom of BD pulmonary involvement, and may be observed in up to 90 % of patients with such involvement [120]. Massive haemoptysis (>500 cc) occurs in about 25–45 % patients and may warrant surgical or instrumental rescue treatments [120]. Other common symptoms are less specific and include cough, fever, dyspnea and pleural chest pain. Fever is of particular interest in BD because it has been shown to be associated with ongoing arterial involvement [102].
Pulmonary Artery Aneurysm
Pulmonary artery aneurysm (PAA) is a major and life-threatening complication of BD. It is well recognized as the most specific pulmonary complication of BD, the second most frequent site of arterial involvement and the leading cause of mortality in BD. [120]. Like any other severe manifestation of BD (eye or neurological involvement), PAA is more frequent in male than female [119]. Prevalence of PAA in the course of BD is not known in the absence of prospective study but ranges from 0.5 to 1 % in retrospective studies [119, 120].
Several Turkish studies [103, 118–121] have helped defining the clinical presentation, prognosis and treatment of PAA. This complication can either occur at diagnosis or during the course of BD. PAA is more frequent than thrombosis of pulmonary artery in BD [120]. It may precede other symptoms of BD, including oral ulcerations, and thus make positive diagnosis of BD difficult. In a cumulative study of PAA cases, almost 14 % of patients did not fulfil ISG criteria [118]. When lacking mucocutaneous or eye lesions, BD is sometimes referred to as Hugues-Stovin syndrome, which associates PAA and deep venous thrombosis [122]. It is still debated whether Hugues-Stovin syndrome is an incomplete phenotype form of BD or a separate nosologic entity [123–125]. Indeed, pulmonary involvement is indistinguishable between the two entities [122].
Pulmonary Artery Thrombosis
Pulmonary artery thrombosis (PAT) is the second most frequent pulmonary manifestation of BD [120], and can be isolated or associated to PAA. Clinical presentation of PAT is not specific and therefore is indistinguishable from common pulmonary embolism or PAA as it includes cough, pleuritic chest pain, fever and dyspnea. Haemoptysis can occur but is significantly less frequent [120] and has been reported to be less abundant [118] in PAT than in PAA.
The term PAT is more commonly used than pulmonary embolism in BD because pulmonary artery occlusion seems to be consecutive to in situ thrombosis rather to thromboembolic mechanisms [126, 127]. Nevertheless, PAT is frequently associated to deep venous thrombosis, and therefore the exact mechanism of pulmonary artery occlusion in BD is still debated [128]. It is important to note that in some patients PAT may transform into PAA, and therefore could occasionally be a forerunner of PAA [120].
Pulmonary Parenchymal Involvement
Parenchymal involvement in BD is commonly associated with pulmonary vascular lesions [120]. Only a few cases of patients with isolated parenchymal lesions have been reported. Clinical presentation of parenchymal involvement in BD is non-specific and includes cough, sputum and chest pain. Differential diagnosis with infection is a major concern in immunocompromised patients [103].
Some authors believe that parenchymal lesions in BD could be small-vessel vasculitis [103], with pulmonary haemorrhage and infarction being the main pathological features [118]. Pathological evaluation of five peripheral lung nodules has recently been reported: in three the lesions comprised both necrosis and pulmonary infarction, in one it was necrotizing granulomatous inflammation, while organizing pneumonia was found in the remaining one [120]. Presence of organizing pneumonia in parenchymal lesions of BD has been reported elsewhere [129]. Thus, two pathogenic mechanisms may explain nodular opacities in BD: those with transient and corticosteroid-responsive nodules may correspond to organizing pneumonia, while slowly-changing nodules evolving to cavitations would be more suggestive of necrosis and infarction [120].
Laboratory Findings
Biological results are not helpful for positive diagnosis of BD and there is no pathognomonic test to date. Laboratory findings are non-specific and are frequently normal. Biological inflammatory syndrome could be suggestive of arterial involvement if infection is ruled out [102]. HLA typing is frequently mentioned but must not be used as diagnostic test as its sensitivity and specificity is low. Furthermore, it is still a matter of debate whether HLA-B51 positive patients have a more severe course of the disease [130]. Therefore, association of HLA B51 to BD is mainly of epidemiological interest.
Imaging Studies
Chest X-ray in BD patients with PAA is often abnormal, showing hilar enlargement and unilateral or bilateral round hilar opacities. Other features associated to PAA are peripheral consolidation consistent with lung infarction, infiltrations related to pulmonary haemorrhage and pleural effusion. Contrary to PAA, chest X-ray in PAT is mostly normal and if abnormal mostly shows only non-specific changes such as pleural effusion and consolidations.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


