IMPORTANT TOPICS
HIGH-RESOLUTION COMPUTED TOMOGRAPHY FINDINGS OF PULMONARY VASCULAR DISEASE
Small Vessel Vasculitis Associated with Antineutrophilic Cytoplasmic Antibody
Idiopathic Pulmonary Hemosiderosis
Abbreviations Used in This Chapter | |
ACS | acute chest syndrome |
ANCA | antineutrophilic cytoplasmic antibody |
BD | Behçet disease |
BMPR2 | bone morphogenetic protein receptor type 2 |
CPTE | chronic pulmonary thromboembolism |
CSS | Churg-Strauss syndrome |
DAH | diffuse alveolar hemorrhage |
FES | fat embolism syndrome |
GPA | granulomatosis with polyangiitis |
HIV | human immunodeficiency virus |
HPAH | heritable pulmonary arterial hypertension |
HPS | hepatopulmonary syndrome |
ILD | interstitial lung disease |
IPAH | idiopathic pulmonary arterial hypertension |
IPH | idiopathic pulmonary hemosiderosis |
MPA | microscopic polyangiitis |
PA | pulmonary artery |
PAH | pulmonary arterial hypertension |
PAN | polyarteritis nodosa |
PAP | pulmonary artery pressure |
PCH | pulmonary capillary hemangiomatosis |
PH | pulmonary hypertension |
PVD | pulmonary vascular disease |
PVOD | pulmonary veno-occlusive disease |
PVRi | pulmonary vascular resistance index |
Pulmonary hypertension (PH) and pulmonary vascular disease (PVD) are often associated with nonspecific symptoms of respiratory dysfunction and nonspecific pulmonary function test findings. PH may result from cardiac or pulmonary abnormalities, or vascular diseases primarily affecting the small arteries or veins. In most cases, PVD and PH are assessed using techniques and imaging modalities other than high-resolution computed tomography (HRCT). However, in some patients, HRCT may be performed to determine whether lung disease is present as a cause of the patient’s disability or to evaluate specific small vessel diseases. If vascular disease is suspected, assessment of the lung using multidetector-row spiral HRCT with contrast infusion is an ideal technique. Furthermore, in patients who have known PH, HRCT may be performed when the etiology of the vascular disease is unclear. Only those vascular diseases commonly assessed using HRCT are described in this chapter.
HIGH-RESOLUTION COMPUTED TOMOGRAPHY FINDINGS OF PULMONARY VASCULAR DISEASE
PH and PVD may be associated with a number of findings on HRCT (1–3). These include alterations in the size of large or small pulmonary arteries; mosaic perfusion, commonly seen in patients who have pulmonary vascular obstruction of various causes; findings of pulmonary edema and hemorrhage, including ground-glass opacity, consolidation, or interlobular septal thickening; centrilobular nodular opacities; and cardiac abnormalities.
Pulmonary Artery Abnormalities
Of primary importance in making the diagnosis of PVD is the recognition of increased or decreased pulmonary artery (PA) diameter. In most cases, the diameters of main, right and left, and intrapulmonary artery branches can be determined on HRCT obtained without contrast infusion, because these arteries are usually outlined by mediastinal fat or air-containing lung.
Increased Artery Diameter
Dilatation of the main PA usually indicates the presence of PH and can be an important finding in recognizing the presence of PVD on HRCT (Fig. 22-1). Even when HRCT scans are obtained at 2-cm intervals, at least one image traverses the main PA, allowing its measurement on soft-tissue window scans.
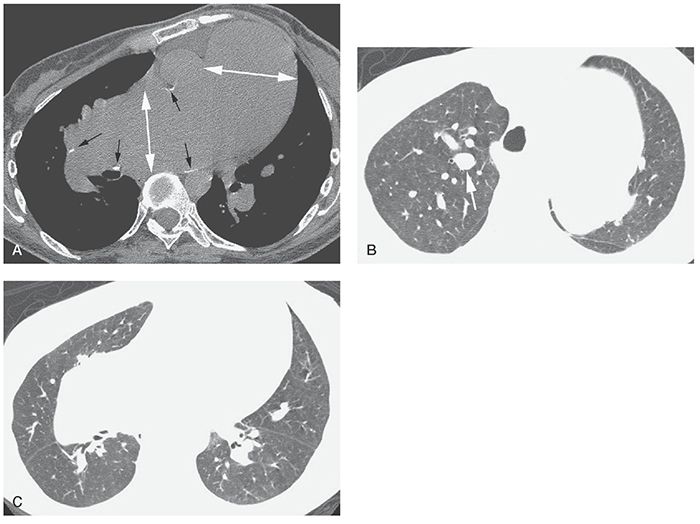
FIGURE 22-1 A–C: PH in a patient with Eisenmenger syndrome associated with congenital heart disease. HRCT was performed to exclude lung disease. A: Marked dilatation of the main pulmonary artery and right pulmonary artery is visible (white arrows). The main pulmonary artery is significantly larger than the aorta. Calcification of the artery wall (black arrows) reflects atherosclerosis. B: Enlargement of central pulmonary artery branches is also visible (arrow). C: Despite marked enlargement of central arteries, peripheral pulmonary vessels appear normal or reduced in diameter.
The main PA measures up to 30 mm in diameter in normal subjects. This measurement is best made at a right angle to the long axis of the PA, lateral to the ascending aorta, and near the level of its bifurcation. In a CT study of normal subjects by Guthaner et al. (4), the main PA diameter (PAD) was found to average 28 ± 3 mm. Kuriyama et al. (5) measured PADs in both normal subjects and patients who have PH. In normal subjects, the main PA averaged 24.2 ± 2.2 mm in diameter at a level near its bifurcation (5). Based on these data, the authors concluded that 28.6 mm (mean plus 2 standard deviations) should be considered the upper limit of normal for main PAD; this value was found to accurately distinguish patients who have PH from normal subjects. Also, Kuriyama et al. (5) found that the main PAD correlated well with pulmonary artery pressure (PAP).
In a study of patients who had chronic lung disease or PVD (6) who were awaiting lung or heart-lung transplantation, considerably more overlap in PAD was seen between patients who had normal or elevated PAPs. Main PAD measured 28 ± 7 mm in patients having normal PAP (≤18 mm Hg) and 33 ± 11 mm in those who have PH (pressure > 18 mm Hg).
The diameter of the main PA may also be compared with that of the aorta; this is quickly and easily done on HRCT. In normals, the PA is usually smaller than the adjacent aorta. In a study by Ng et al. (7), the ratio of the diameter of the main PA to the aortic diameter (A) was measured using HRCT in 50 patients who had a variety of pulmonary and cardiovascular diseases, and also had PAPs measured at right heart catheterization. Measurement of vessel diameters was made at the level at which the right PA traverses the mediastinum. Both the diameters of the PA and the PA/A ratio were significantly related to PAP (r = 0.74, p < 0.0005) (7). For patients younger than 50 years, PAP correlated more closely with PA/A (r = 0.77, p < 0.00005) than with PAD (r = 0.59, p < 0.005); for patients older than 50 years, the opposite was true. More important, a PAD to aortic diameter ratio of more than 1 strongly suggests PH (Fig. 22-1A). In this study (7), the specificity and positive predictive value of this finding were 92% and 96%, respectively. The sensitivity and negative predictive value were lower, measuring 70% and 52%, respectively. Thus, a PA/A of less than 1 does not necessarily mean PAP is normal.
Devaraj et al. (8) assessed the accuracy of CT and echocardiographic measurements in predicting the presence of PH in 77 patients. The ratios of the diameter of the main pulmonary artery to the diameter of the ascending aorta and of the cross-sectional area of the pulmonary artery to the diameter of the ascending aorta (r2 = 0.45, p < 0.001) correlated equally with mean pulmonary artery pressure (mPAP). The ratio of the diameter of the main pulmonary artery to the diameter of the thoracic vertebra, the segmental arterial diameter, and the segmental artery-to-bronchus ratio were related to mPAP, but did not strengthen correlations compared with the ratio of the diameter of the main pulmonary artery to the diameter of the ascending aorta alone.
Dilatation of main pulmonary arteries may also be seen in the presence of PH. The right and left pulmonary arteries should be of approximately equal size, although the left PA appears slightly larger in most subjects. In the study by Kuriyama et al. (5), the proximal right PA measured 18.7 ± 2.8 mm in diameter in normals, and the left PA averaged 21.0 ± 3.5 mm. In the study by Ackman Haimovici et al. (6) of transplantation patients, the left PA averaged 21 ± 5 mm in those who have normal PAP.
Pulmonary artery aneurysms are rare. They may be posttraumatic, mycotic, or related to some vasculitis syndromes. Pulmonary artery aneurysms may be seen in Behçet disease (BD); Hughes-Stovin syndrome, which is likely a variant of BD and rarely in giant cell arteritis (9–11).
Within the lung, the diameter of a small PA and its neighboring bronchus should be approximately equal, although vessels usually appear slightly larger than their accompanying bronchus, particularly in dependent lung regions. In patients who have PH or increased blood volume or blood flow (12), significant dilatation of these small vessels relative to adjacent bronchi may be seen (Fig. 22-1B) (13).
Focal dilatation of peripheral pulmonary arteries may be seen in patients with acute or chronic pulmonary embolism, owing to impaction of clot within the vessel. A similar phenomenon may be seen in patients with tumor embolism or other causes of nonthrombotic pulmonary embolism (11,14,15). Arteries in the lung periphery appear irregularly dilated and may have a beaded or varicose appearance in patients with tumor embolization (11). This may mimic the appearance of tree-in-bud (16,17). Dilatation of small vessels in the lung periphery may also be seen in hepatopulmonary syndrome (HPS) (13), pregnancy (11,18), in patients with multiple small pulmonary arteriovenous fistulas (11), and in some cardiac malformations.
Decreased Artery Diameter
Decreased diameter of some intrapulmonary arteries is common in patients who have PH or regional decrease in pulmonary blood flow associated with large or small artery disease (Fig. 22-2). This abnormality is usually recognized in association with inhomogeneous lung attenuation (i.e., mosaic perfusion) (Fig. 22-2C,D) (2,19–21). An abrupt decrease in size of a PA along its course, visible on lung window scans, is suggestive of chronic pulmonary thromboembolism (CPTE) as the cause of PH or PVD. Asymmetry in the size of pulmonary arteries in the right and left lungs visible on lung window scans may also be seen in patients who have CPTE.
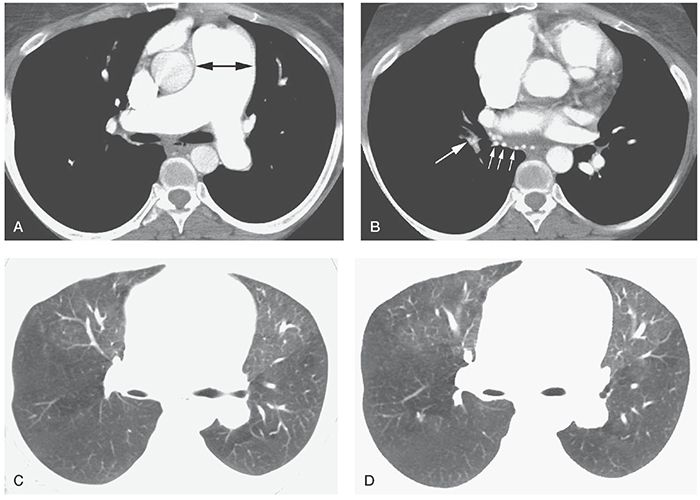
FIGURE 22-2 A–D: Chronic pulmonary embolism. A: On a contrast-enhanced multidetector-row spiral HRCT, marked enlargement of the main pulmonary artery (arrow) and right pulmonary artery is present. B: The right interlobular pulmonary artery (large arrow) is reduced in diameter compared with the left side, and bronchial artery dilatation is present (small arrows). C and D: Lung window scans show reduced vessel size in the posterior lungs, particularly on the right, associated with reduced lung attenuation (i.e., mosaic perfusion). The patient was treated using embolectomy.
Narrowing of central pulmonary arteries with thickening of their walls, localized regions of stenosis, and poststenotic dilatation may be seen in patients with Takayasu arteritis or giant cell arteritis (9,11).
Pulmonary Artery Obstruction
Obstruction of large or small PA branches by thrombus or nonthrombotic emboli occurs in several abnormalities, most commonly acute or CPTE (22–24), but also including pulmonary artery sarcoma (25) and tumor emboli (14,15). Current multidetector spiral CT scanners allow contrast-enhanced volumetric HRCT (11) for the detailed assessment of both pulmonary arteries and the lung parenchyma.
Mosaic Perfusion and Mosaic Lung Attenuation
Mosaic perfusion refers to inhomogeneous lung attenuation resulting from inhomogeneous blood flow (2,11,19–21). It may result from vascular disease (e.g., CPTE, vasculitis) or airways disease (see Chapter 7). Mosaic perfusion is commonly associated with decreased size of pulmonary vessels within relatively lucent lung regions (Figs. 22-2 and 22-3). If the combination of decreased vessel size and decreased lung attenuation is visible, a diagnosis of mosaic perfusion is easily made.
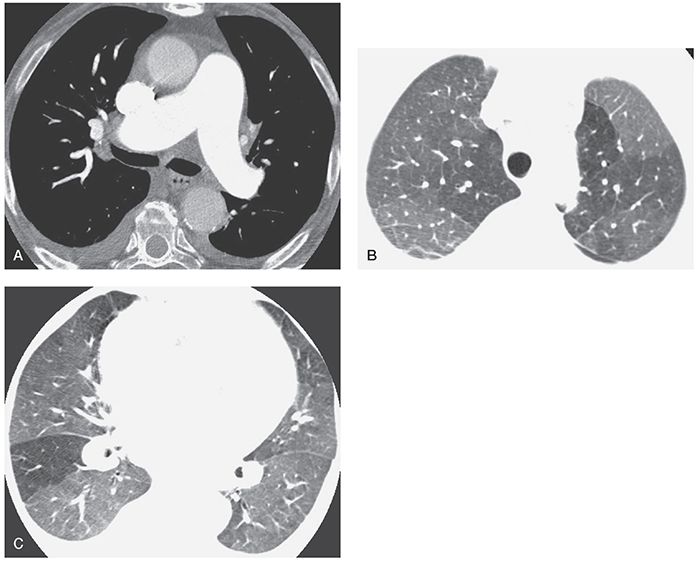
FIGURE 22-3 A–C: PH and mosaic perfusion in a patient with chronic pulmonary embolism. A: On a contrast-enhanced multidetector-row spiral HRCT obtained using 1.25-mm detector width, the main pulmonary arteries appear dilated. B and C: At lung window settings, areas of decreased opacity reflect mosaic perfusion. Vessels within the lucent regions are reduced in size.
In a study of pulmonary parenchymal abnormalities in 75 patients with CPTE, 58 (77.3%) patients showed typical findings of mosaic perfusion with normal or dilated arteries in areas of increased attenuation (26). In this study, areas of relatively increased attenuation averaged –727 HU, whereas areas of decreased attenuation averaged –868 HU. In another study of patients with PH due to CPTE, PH of other causes, and a variety of other pulmonary diseases, HRCT was believed to show mosaic perfusion in all patients with CPTE (27). Considerably more variation in vessel size in different lung regions was also visible in the patients with CPTE. Overall, HRCT had a sensitivity of 94% to 100% and a specificity of 96% to 98% in making the diagnosis of CPTE (27). Mosaic perfusion is less frequent in patients with acute pulmonary embolism, but may be seen (28–30). Patients with large vessel vasculitis resulting in pulmonary artery stenosis may also show this finding. Such diseases include Takayasu arteritis and giant cell arteritis (9).
Some patients who have PVD may show perfusion abnormalities without a clear-cut decrease in vessel size or may show patchy ground-glass opacity that mimics the appearance of mosaic perfusion. If it is unclear whether inhomogeneous lung attenuation represents mosaic perfusion, the terms mosaic lung attenuation or mosaic pattern may be used (19,21,31).
In patients who have PH, mosaic lung attenuation is seen significantly more often in patients who have PH due to vascular disease than in patients who have PH due to cardiac or lung disease, and CPTE is the most common disease responsible for this finding. The frequency with which mosaic lung attenuation is seen on CT in patients who have various causes of PH has been studied by Sherrick et al. (19). In this study, 23 patients had PH due to vascular disease, 17 patients had PH due to cardiac disease, and 21 patients had PH due to lung disease. Of the 23 patients with PH due to vascular disease, 17 (74%) had mosaic lung attenuation. Twelve of the 17 patients with mosaic lung attenuation due to vascular disease had CPTE, 2 had idiopathic or primary PH, 2 had pulmonary veno-occlusive disease (PVOD), and 1 had fibrosing mediastinitis associated with vascular obstruction. Among the 17 patients with PH due to cardiac disease, only 2 (12%) patients had a mosaic pattern of lung attenuation (19). Of the 21 patients with PH due to lung disease, 1 (5%) patient had mosaic lung attenuation.
Abnormalities of pulmonary perfusion in patients with PH do not always result in mosaic perfusion recognizable on HRCT. In a study of five patients with scleroderma, normal lung parenchyma, and PH, HRCT showed a reduction in the anterior to posterior lung attenuation gradient, when compared to patients without PH (2,32). This is perhaps related to reduced compliance of the pulmonary vasculature in the patients with PH.
High-Resolution Computed Tomography Differentiation of Causes of Mosaic Attenuation
On HRCT, it is often possible to distinguish among ground-glass opacity, mosaic perfusion caused by airways disease, and mosaic perfusion caused by vascular disease. In two studies (20,33), an accurate distinction was possible in more than 80% of cases based on HRCT findings. However, findings may be nonspecific or misleading in some cases.
In patients with mosaic perfusion resulting from airways disease, the presence of airway abnormalities in lucent lung regions may allow the correct diagnosis. Abnormal dilated or thick-walled airways (i.e., bronchiectasis) are visible in approximately 70% of patients with mosaic perfusion related to airways disease (34–38). It is important to note, however, that dilatation of segmental and subsegmental bronchi has also been reported in some patients with CPTE (39). In a study by Remy-Jardin et al. (39), cylindrical bronchial dilatation was found in 21 of 33 (64%) patients with CPTE, and bronchial wall thickening was identified in 4 (12%).
Lobular areas of lucency (i.e., mosaic attenuation with a lobular pattern) are more common in patients with airways disease than in vascular disease. In a study by Im et al. (40) of 48 consecutive patients with lobular areas of low attenuation seen on HRCT, 46 (95%) had symptoms related to respiratory disease, and only 2 patients had vascular disease.
In patients with pulmonary vascular obstruction (e.g., CPTE) as a cause of mosaic perfusion, dilatation of central pulmonary arteries may be present as a result of PH, and areas of low attenuation are usually larger than pulmonary lobules (e.g., segments or lobes).
Ground-glass opacity may be accurately diagnosed as the cause of inhomogeneous lung opacity if it is associated with other findings of infiltrative disease such as consolidation, reticular opacities, honeycombing, or nodules. Ground-glass opacity may also result in very ill-defined and poorly marginated areas of increased opacity, lacking the sharply marginated and geographic appearance sometimes seen in patients with mosaic perfusion. Ground-glass opacity can often be diagnosed simply because lung looks too dense, although this is quite subjective and depends on using consistent window settings and being familiar with the appearance of normal lung parenchyma.
Expiratory HRCT scans may be useful in the diagnosis of mosaic attenuation and often allow the differentiation of mosaic perfusion resulting from airways obstruction from other abnormalities when inspiratory scans are inconclusive. In patients with ground-glass opacity, expiratory HRCT typically shows a proportional increase in attenuation in areas of both increased and decreased opacity. In patients with mosaic perfusion resulting from airways disease, attenuation differences are accentuated on expiration; relatively dense areas increase in attenuation, whereas lower attenuation regions remain lucent (i.e., air trapping is present) (see Chapter 3) (34,41–43).
In a study by Arakawa et al. (20) of patients showing mosaic attenuation as their predominant HRCT abnormality, the accuracy of HRCT in correctly diagnosing the type of disease present increased from 81% to 89% in patients with ground-glass opacity and from 84% to 100% in diagnosing airways disease when expiratory scans were included in the analysis (20). Some patients who appear to show ground-glass opacity on inspiratory scans and show air trapping on expiratory scans may thus be correctly diagnosed as having obstructive disease.
In patients with mosaic perfusion resulting from vascular disease, air trapping is not usually seen, and expiratory HRCT findings often mimic those seen in patients with ground-glass opacity. However, in a study of patients with inhomogeneous lung attenuation of various causes (33), air trapping was believed to be present on expiratory scans in some patients with vascular disease when scans were viewed blindly. Furthermore, air trapping has been reported in patients with acute pulmonary embolism, likely due to hypoxemic bronchoconstriction (29). In 15 patients with pulmonary embolism studied by Arakawa et al. (29), mosaic perfusion was identified in 7 (46.7%) patients and air trapping in 9 (60%) patients. Of 32 areas of mosaic perfusion identified, 23 (71.9%) showed air trapping on expiratory scans.
Findings of Pulmonary Edema and Hemorrhage
Pulmonary edema may be associated with various PVDs, including some causes of PH and pulmonary vasculitis (2,44–46). It may be manifested by centrilobular, patchy, or diffuse ground-glass opacity, consolidation, interlobular septal thickening, peribronchovascular interstitial thickening, or a combination of these.
Pulmonary hemorrhage may also result in diffuse pulmonary abnormalities, and is common in pulmonary vasculitis syndromes. It may appear as centrilobular or diffuse ground-glass opacity, consolidation, or sometimes associated with interlobular septal thickening (46). Pulmonary edema and pulmonary hemorrhage are discussed in Chapter 18.
Pulmonary infarction resulting from vascular disease typically shows focal areas of consolidation, often wedge shaped and peripheral, and may be associated with a feeding vessel, surrounding ground-glass opacity (i.e., the “halo sign”), central lucencies, and absent air bronchograms (47,48). In a recent study (49), CT findings occurring with a higher frequency with pulmonary infarction than with other causes of consolidation included a feeding vessel (32% vs. 11%, respectively; p = 0.029) and central lucencies (46% vs. 2% respectively; p < 0.001). Air bronchograms, in contrast, were seen in a lower percentage of patients with pulmonary infarction (8% vs. 40%, respectively; p = 0.003). Localized nodular or masslike opacities, with or without cavitation, may also be seen in vasculitis syndromes, particularly granulomatosis with polyangiitis (GPA) (Wegener granulomatosis) and Churg-Strauss granulomatosis (50,51).
Centrilobular Opacities and Nodules
In patients with PH, ill-defined centrilobular opacities may be seen in patients who have plexogenic angiopathy (52), capillary pulmonary hemangiomatosis associated with proliferation of small vessels (53,54), PVOD with pulmonary edema, lobular mosaic perfusion, pulmonary hemorrhage, or cholesterol granulomas likely related to repeated episodes of hemorrhage (2,55).
Processes resulting in a vascular and perivascular inflammation, including vasculitis (56) and reaction to injected substances, such as talc (57–59), can produce ill-defined centrilobular opacities visible on HRCT. Connolly et al. (56) reported hazy or fluffy centrilobular, perivascular opacities in eight children with vasculitis, including five with GPA (Wegener granulomatosis), one with systemic lupus erythematosus, one with scleroderma-polymyositis overlap syndrome, and one with Churg-Strauss syndrome (CSS). In these eight children, centrilobular opacities were associated with the onset of active disease or an exacerbation of preexisting disease. In four of five patients, this abnormality disappeared on treatment.
Cardiovascular Abnormalities Associated with Pulmonary Hypertension
In addition to enlargement of the pulmonary arteries, a number of cardiovascular abnormalities visible using CT have been associated with PH, and many of these are related to right ventricular dysfunction and failure. These findings are largely beyond the scope of this book and are principally used in the assessment of patients with acute pulmonary embolism. Nonetheless, some useful findings are reviewed briefly.
Enlargement of the right ventricle and right atrium is common in patients who have PH, being seen in all patients who had PH studied by Bergin et al. (27). Findings of right atrial or right ventricular enlargement with flattening of the ventricular septum, or septal bowing to the left, are valuable in the diagnosis of PH, but require contrast infusion. Additional findings of PH include dilatation of the inferior vena cava (23,27), contrast reflux into the inferior vena cava (60), measurement of right ventricular (RV) and left ventricular (LV) short axes, RV/LV short-axis ratio, and superior vena cava and azygos vein diameters (61,62). Pericardial thickening or effusion may also be present. ECG-gated MDCT can be useful in the identification of cardiac abnormalities associated with PH (63).
PULMONARY HYPERTENSION
PH is defined as an abnormal elevation of pressure in pulmonary circulation, with a mean pulmonary arterial pressure higher than 25 mm Hg, regardless of the underlying mechanism. The clinical classification system for PH was updated at the Fourth World Symposium on Pulmonary Hypertension in Dana Point, California, in 2008 (63,64).
PH may have a variety of causes, and its treatment may vary considerably depending on its etiology (65,66). The treatment of PH in different diseases may require pharmacologic intervention with the use of anticoagulants or vasodilators, embolectomy, or lung transplantation (66).
The assessment of patients who have PH usually involves techniques other than HRCT (63–66). However, HRCT may be performed (a) to assess patients who have PH related to lung disease (i.e., emphysema or pulmonary fibrosis), (b) in patients having PH of unknown cause, (c) in patients believed to have vasculitis or small vessel disease, or (d) in patients being evaluated for lung transplantation. Also, patients who have PH occurring because of CPTE often have CT for diagnosis.
The term primary pulmonary hypertension has been used to refer to PH of obscure cause or, more specifically, to idiopathic PH associated with plexogenic arteriopathy (66). PH associated with a specific disease has been referred to as secondary pulmonary hypertension. Secondary PH is most commonly associated with lung disease (e.g., emphysema, chronic obstructive pulmonary disease (COPD), idiopathic pulmonary fibrosis) and cardiac disease (e.g., chronic left-to-right shunts, left heart failure, mitral valve lesions), but other diseases (e.g., drug use, embolism of foreign material, obesity, sleep apnea, human immunodeficiency virus [HIV] infection, pericardial disease) may also result in PH (2,11,19,66).
In 2003, the clinical classification of PH was revised by the Third World Symposium on Pulmonary Arterial Hypertension held in Venice (67). In this classification, the terms “primary PH” and “secondary PH” were discarded in favor of a more precise description based on etiology, histologic findings, and associated diseases (67). Subsequently, at the Fourth World Symposium on PH held in 2008 in Dana Point, California, the consensus of an international group of experts was to maintain the general philosophy and organization of the Venice classifications. However, in response to a questionnaire regarding the previous classification, a majority of experts (63%) felt that modification of the Venice classification was required to accurately reflect information published over the past 5 years, as well as to clarify some areas that were unclear (68,69).
In the Dana Point classification (Table 22-1), a large category termed “pulmonary arterial hypertension (PAH)” is divided into (a) idiopathic pulmonary arterial hypertension (IPAH), (b) heritable pulmonary arterial hypertension (HPAH), (c) PAH associated with drugs or toxins, (d) PAH associated with collagen-vascular disease, HIV, portal hypertension, congenital heart disease, schistosomiasis, or chronic hemolytic anemia, and (e) PAH associated with venous or capillary abnormalities, specifically PVOD and pulmonary capillary hemangiomatosis (PCH), presenting in a similar fashion and with similar histologic abnormalities of small pulmonary arteries, including intimal fibrosis, medial hypertrophy, and plexiform arteriopathy. This classification also recognizes separate categories for PH with left heart disease, PH associated with lung disease and/or hypoxemia, PH due to chronic thrombotic and/or embolic disease, and PH with unclear multifactorial mechanisms (Table 22-1).
TABLE 22-1 Clinical Classification of Pulmonary Hypertension (DANA POINT, 2008)

Pulmonary Arterial Hypertension
The diseases described using this term are characterized, at least in part, by plexogenic arteriopathy, a histologic abnormality consisting of a disorganized proliferation of small muscular arteries, endothelial cells, smooth muscle cells, and myofibroblasts (i.e., a plexiform lesion) (Fig. 22-4) (66). This abnormality has also been referred to as pulmonary hypertensive arteriopathy (70,71). As indicated previously, PAH may be idiopathic, familial, associated with various conditions, or associated with significant venous or capillary abnormalities (PVOD and PCH). Many of the causes of PAH result in similar HRCT findings, although PVOC and PCH show additional findings on HRCT that generally result in a distinct appearance.
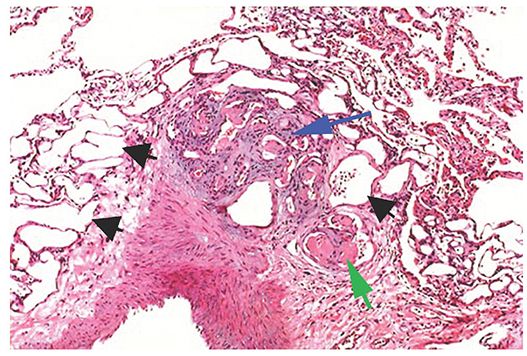
FIGURE 22-4 Plexiform arteriopathy in PAH. At the center of the picture is a compact mass of angioblastic tissue or granulation tissue (i.e., a plexiform lesion, blue arrow), originating from a large muscular artery at lower left. Endothelial cells are prominent. Numerous empty vascular channels are visible (black arrow). Some vascular lumens contain fibrin thrombi (green arrow). (Courtesy of Martha Warnock, MD.)
Idiopathic Pulmonary Arterial Hypertension
IPAH is most common in patients 30 to 40 years of age, and women are affected more often than men. Symptoms include the insidious onset of dyspnea on exertion. Progressive symptoms, cor pulmonale, and death within a few years are typical (72). Pathologic abnormalities principally involve muscular pulmonary arteries (less than 1 mm in diameter) (67).
Enlargement of the central pulmonary arteries is common in patients with IPAH (Table 22-2) (11). In an HRCT study of 15 patients with IPAH (73), the main pulmonary artery was larger than the aorta in 93%. Mosaic attenuation may be seen, but is not a conspicuous feature of this disease, as it is in patients with CPTE (2,19). In one study, HRCT in five patients with IPAH showed enlargement of central pulmonary arteries, normal lung parenchyma (n = 3), and mosaic lung attenuation (n = 2) (Figs. 22-5 and 22-6). Interlobular septal thickening is visible in about 10% of patients (73). Cardiomegaly was visible in all 15 patients with IPAH in one study (67). Pericardial and pleural effusions were seen in 60% and 13%, respectively (73).
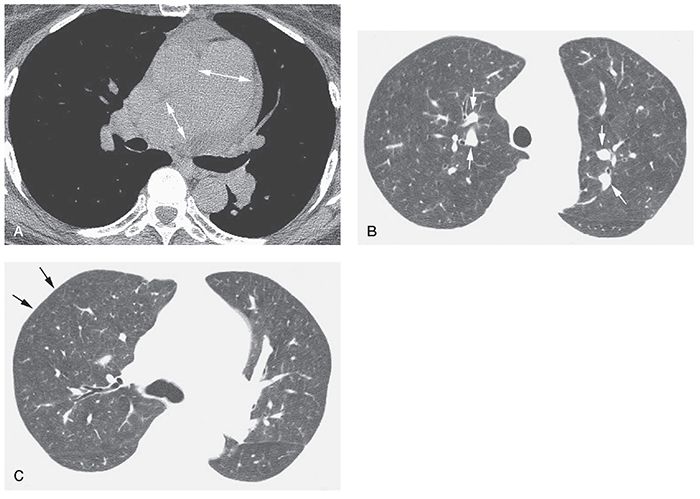
FIGURE 22-5 A–C: Idiopathic PH with plexogenic arteriopathy. A: Enlargement of the main pulmonary artery and right pulmonary arteries is visible at tissue windows (arrows). A small pericardial effusion is present. B: At a lung window, central pulmonary artery branches (arrows) are increased in diameter and appear much larger than their adjacent bronchi. C: Pulmonary arteries appear relatively small in the lung periphery (arrows). Lung parenchyma shows a slightly inhomogeneous attenuation, likely due to mosaic perfusion.
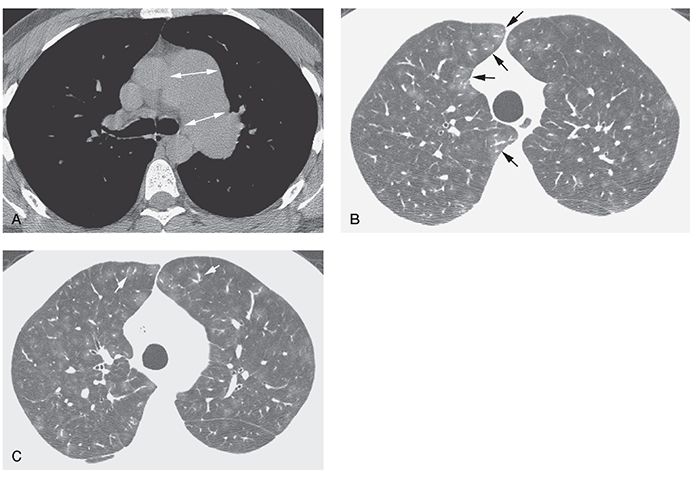
FIGURE 22-6 A–C: Idiopathic PH with plexogenic arteriopathy. A: Enlargement of the main and left pulmonary artery is visible at tissue windows (arrows). B and C: Lung window scans through the upper lobes show inhomogeneous lung attenuation with a lobular or centrilobular distribution. Relatively large vessel size in areas of increased attenuation (arrows) suggests mosaic perfusion as the cause of inhomogeneous opacity. A similar appearance was visible in the lower lobes. Histologic examination of the lungs after removal for lung transplantation showed plexogenic arteriopathy.
TABLE 22-2 HRCT Findings in Idiopathic Pulmonary Arterial Hypertension
Symmetric dilatation of the central pulmonary arteriesa |
Mosaic perfusion |
Ground-glass opacities |
Ill-defined centrilobular ground-glass opacities or nodules |
Interlobular septal thickening |
aMost common findings.
Ill-defined centrilobular opacities are sometimes seen in patients who have IPAH (11). In a review of 15 patients with IPAH (67), ground-glass opacities were visible in 33%, and in most of those, the opacities were centrilobular in location. This finding may represent patchy mosaic perfusion or the presence of plexogenic lesions (52), or may be the result of hemorrhage (Fig. 22-6). Also, cholesterol granulomas may result in small ill-defined centrilobular nodules in patients who have PH. In one study, histopathologic evidence of cholesterol granulomas was found in 5 of 20 (25%) patients with severe PH, and 2 of these 5 had IPAH (55). In 3 of these 5 patients, the granulomas were visible on HRCT as small centrilobular nodules. Cholesterol granulomas may result from repeated pulmonary hemorrhage with macrophage ingestion of red blood cells (2).
Heritable Pulmonary Arterial Hypertension
The primary genetic defect associated with HPAH, identifiable in more than 70% of cases, is a mutation in the gene encoding bone morphogenetic protein receptor type 2 (BMPR2), a member of the transforming growth factor beta superfamily; other mutations are more rarely associated. BMPR2 mutations also occur in 10% to 40% of apparently isolated cases of IPAH (68,74,75). IPAH and HPAH have similar clinical, functional, and survival characteristics (75).
Drug and Toxin-Induced PAH
PAH has a clear association with the use of various drugs and toxins use (52,67,68), including fenfluramine and dexfenfluramine taken for weight loss (Fig. 22-7). Clinical, functional, and hemodynamic features are similar to IPAH.
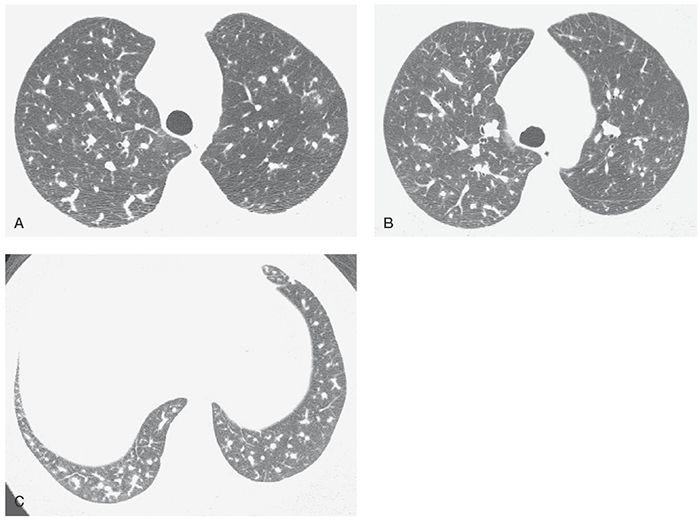
FIGURE 22-7 A–C: PH associated with use of anorectic drugs. Enlargement of the main pulmonary artery is associated with marked variation in the size of small pulmonary arteries. In general, arteries in the right lung appear larger than the left.
PAH Associated with Other Diseases
PAH occurring in the absence of lung disease may be seen in patients who have collagen-vascular disorders such as scleroderma, mixed connective tissue disease, rheumatoid disease, and systemic lupus erythematosus (76,77). The association is most clearly documented for patients with scleroderma. Generally, the histologic, CT, and HRCT findings are similar to those of IPAH. However, in patients with scleroderma and PAH, the prognosis is markedly worse than in patients with IPAH (68).
PAH is also associated infection with HIV (78–80), liver disease with portal hypertension (portopulmonary hypertension) (81,82), congenital heart disease, schistosomiasis, and chronic hemolytic anemia (68).
Pulmonary Veno-Occlusive Disease and/or Pulmonary Capillary Hemangiomatosis
PVOD and PCH are uncommon, but increasingly recognized as causes of PH (68). PVOD and PCH are quite similar in terms of lung abnormalities, pathologic features, clinical presentation, and associations.
Pulmonary Veno-Occlusive Disease
PVOD is a rare disorder in which gradual obliteration of the pulmonary veins by intimal thickening and fibrosis leads to PAH (83–86). Interlobular or small postcapillary veins may be involved (Fig. 22-8), and the process may involve the lung in a diffuse or patchy fashion (85). Venous obstruction leads to edema of the interlobular septa, septal lymphatic dilatation, and venous infarcts. Patchy dilatation and proliferation of alveolar capillaries are associated with interstitial fibrosis and hemorrhage, and lead to secondary hyperplasia of muscular pulmonary arteries (85).
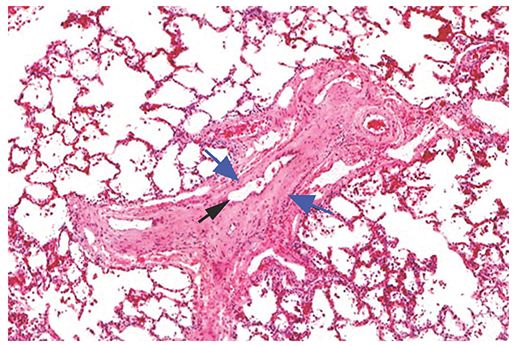
FIGURE 22-8 Histologic appearance of PVOD. Veins within interlobular septa or venules are narrowed or occluded by cellular intimal thickening or hyaline connective tissue (blue arrows). A small residual lumen (black arrow) suggests recanalization of thrombi. Associated pulmonary arteries show some medial and intimal thickening. (Courtesy of Martha Warnock, MD.)
PVOD is typically idiopathic, but has multiple associations, including viral infections, inhaled toxins, deposition of immune complexes in patients with collagen-vascular diseases, Langerhans histiocytosis, a genetic predisposition, AIDS, and use of contraceptive or cytotoxic chemotherapeutic agents. In addition, radiation injury has been proposed as a possible cause. It may affect any age, but it is most common in children and young adults. Symptoms are nonspecific and consistent with PH. Characteristic of PVOD and PCH is elevated PAP but normal or low pulmonary capillary wedge pressure (85). The disease is generally fatal within a few years (85).
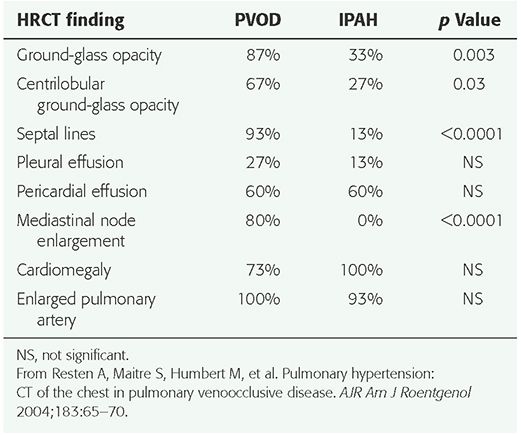
HRCT findings have been described in a number of patients (2,11,53,73,83,85,87,88). The most common findings include (a) smooth interlobular septal thickening (Fig. 22-9); (b) diffuse or multifocal regions of ground-glass opacity, which may be geographic and patchy, perihilar or peripheral, or centrilobular (Fig. 22-10); (c) pericardial or pleural effusions; (d) enlarged central pulmonary arteries; and (e) pulmonary veins of normal caliber; this combination of findings is highly suggestive of PVOD (Table 22-3) (87). In the one study (87), 7 of 8 patients had interlobular septal thickening, and all 8 patients had regions of ground-glass opacity. In a study of 15 patients with pathologically proven PVOD, 93% showed interlobular septal thickening, and 87% showed ground-glass opacity. Centrilobular ground-glass opacities were visible in 67% of the 15 patients, whereas 33% showed panlobular opacities (73). In comparing the HRCT findings in patients with PVOD to those of IPAH, ground-glass opacities (p = 0.003), centrilobular ground-glass opacities (p = 0.03), interlobular septal thickening (p < 0.0001), and mediastinal lymph node enlargement (p < 0.0001) were significantly more frequent in the patients with PVOD (Table 22-4) (73). Histologic correlation with CT findings (87) has shown that thickened interlobular septa corresponded to the presence of septal fibrosis and venous sclerosis. Ground-glass opacity may be related to alveolar wall thickening or pulmonary edema.
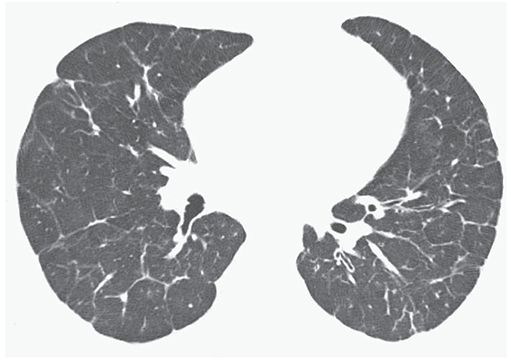
FIGURE 22-9 PVOD in a 21-year-old man. HRCT near the lung bases shows interlobular septal thickening to be a predominant finding.
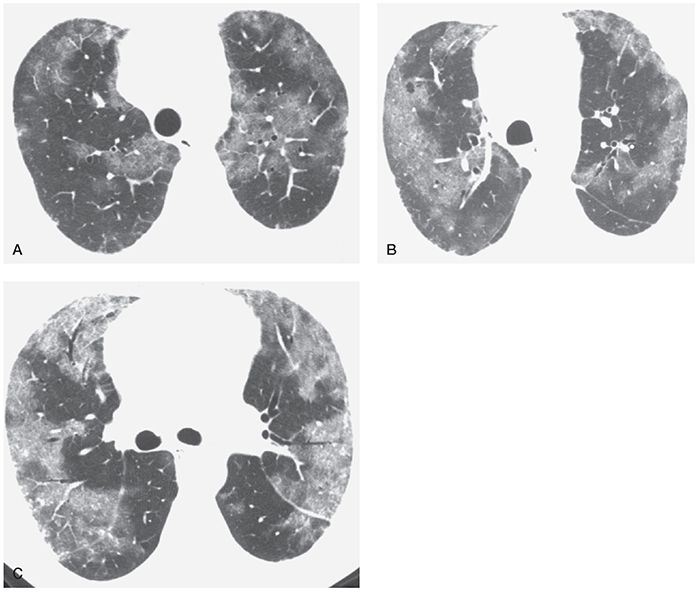
FIGURE 22-10 A–C: Pulmonary veno-occlusive disease. HRCT at three levels shows patchy areas of ground-glass opacity, with a predominantly peripheral distribution. Some intralobular interstitial thickening is visible in the areas of ground-glass opacity, but interlobular septal thickening is not clearly visible.
TABLE 22-3 HRCT Findings in Pulmonary Veno-Occlusive Disease
Symmetric dilatation of the central pulmonary arteriesa |
Interlobular septal thickeninga,b |
Ground-glass opacitiesa,b |
Normal size pulmonary veinsa,b |
Combination of the previous four findingsa,b |
Ill-defined centrilobular ground-glass opacities or nodulesa |
Mediastinal lymph node enlargementa,b |
aMost common findings.
bFinding(s) most helpful in differential diagnosis.
TABLE 22-4 Comparison of Frequency of HRCT Findings in Patients with Pulmonary Veno-Occlusive Disease and Idiopathic Pulmonary Arterial Hypertension
Central pulmonary arteries were considered to be enlarged in 7 of 8 in one study (87). In another study, the main pulmonary artery was larger than the aorta in all 15 (73). Mosaic perfusion may be seen, but is not a prominent feature of this disease (87). Pericardial or pleural effusion (73,87) and mediastinal lymph node enlargement may be seen (73). Cardiomegaly is common, and although right-sided chambers are enlarged, the left atrium and ventricle are normal in size (85).
Pulmonary Capillary Hemangiomatosis
PCH is a very rare cause of PAH, most often occurring in young adults and associated with dyspnea and hemoptysis (89). Although slow progression is typical, mean survival is only 3 years. As with PVOD, PCH is idiopathic, but many associations have been reported, including systemic lupus erythematosus, scleroderma, Takayasu arteritis, Kartagener syndrome, hypertrophic cardiomyopathy, and genetic factors (54,85,89). It has been suggested that PCH may occur secondary to PVOD, because both entities share a number of clinical and pathologic characteristics (86).
Pathologically, PCH represents a patchy interstitial proliferation of thin-walled capillary-size blood vessels within alveolar walls. The sheets of vessels surround, compress, and appear to invade the walls of pulmonary veins and are associated with intimal fibrosis, venous occlusion, interstitial edema, and hemorrhage (86,90). Chest radiographs may be normal, except for findings of PAH, or may show small nodular opacities.
HRCT findings have been reported in a few patients who have this disease (85,91). Ill-defined centrilobular nodules of ground-glass opacity, diffuse in distribution, are most typically described, and patchy or lobular areas of ground-glass opacity may also be seen (Figs. 22-11 and 22-12, Table 22-5). HRCT in two patients who had PCH showed mediastinal and hilar lymph node enlargement, enlargement of pulmonary arteries, and pleural effusions (53). In both patients (53) and in one other reported patient (54), HRCT showed smooth thickening of interlobular septa, small ill-defined centrilobular nodules, and focal regions of lobular or centrilobular ground-glass opacity. In comparison to patients with PVOD, thickened interlobular septa were sparse and few in number (85). Pathologic correlation in these cases showed that the centrilobular opacities correlated with proliferations of small capillary-size vessels and intra-alveolar hemosiderin-laden macrophages.
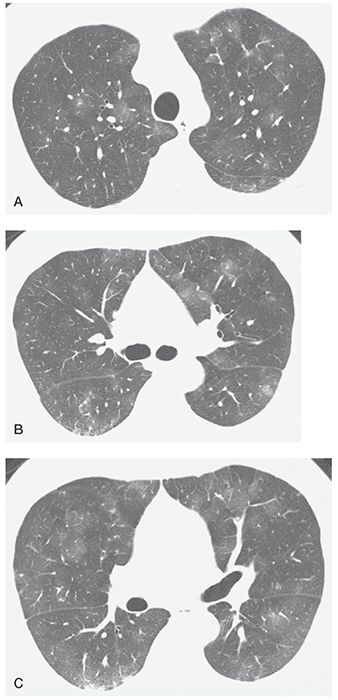
FIGURE 22-11 A–C: Pulmonary capillary hemangiomatosis. HRCT at three levels shows patchy areas of ground-glass opacity, a common finding in this disease. This appearance is nonspecific. The diagnosis was made at open lung biopsy.
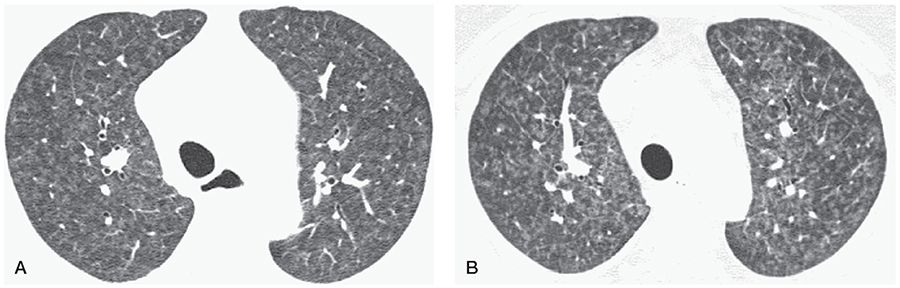
FIGURE 22-12 HRCT in a patient with PH and worsening symptoms after treatment using epoprostenol. A: HRCT shows diffuse centrilobular nodules of ground-glass opacity, suggestive of PCH. B: Four months after beginning treatment, there appears to be progression of nodules, and some interlobular septal thickening is also visible.
TABLE 22-5 HRCT Findings in Pulmonary Capillary Hemangiomatosis
Dilatation of the central pulmonary arteriesa |
Ill-defined centrilobular ground-glass opacities or nodulesa,b |
Lobular ground-glass opacitiesa |
Interlobular septal thickening inconspicuous |
aMost common findings.
bFinding(s) most helpful in differential diagnosis.
Use of High-Resolution Computed Tomography in Determining Treatment in Pulmonary Arterial Hypertension
Therapy commonly used for treatment of patients with PAH can be harmful or even fatal in patients with PVOD or PCH (73,85,88,92). Vasodilators such as prostacyclin and calcium channel blockers are used to treat PAH, and have been shown to increase exercise potential, hemodynamics, and long-term survival. However, these drugs may result in severe pulmonary edema or death in patients with postcapillary PH. In patients with PVOD and PCH, dilation of pulmonary small muscular arteries and arterioles occurring with vasodilator treatment likely results in increased transcapillary hydrostatic pressure and pulmonary edema because of the presence of fixed, elevated pulmonary venous resistance. Lung transplantation remains the most effective means of prolonging survival and improving quality of life for patients with PVOD or PCH (85,88).
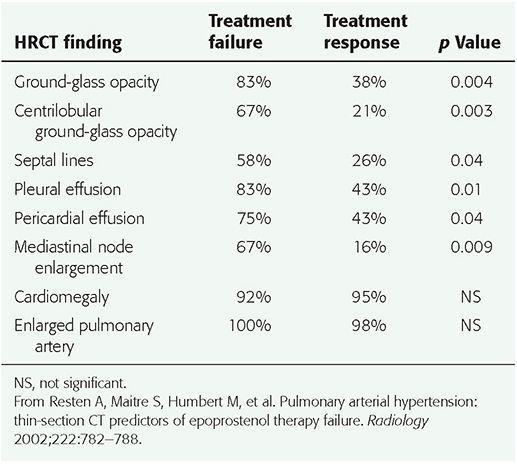
It has been recommended that HRCT be performed prior to vasodilator treatment in patients with PAH. The presence of HRCT findings that suggest the diagnosis of PVOD or PCH, such as interlobular septal thickening or centrilobular ground-glass opacities, should lead to further evaluation before vasodilator treatment is instituted (73,92). In a study by Resten et al. (92), 73 consecutive patients with severe PH treated with epoprostenol (prostacyclin) were retrospectively separated into two groups. Group 1 included 12 patients with epoprostenol therapy failure, leading to death in an average of 1.9 months; 6 of these patients subsequently evaluated at autopsy had PVOD or PCH. The second group of 61 patients improved clinically with epoprostenol; this group was comprised of patients with IPAH, familial PAH, and PAH related to drugs, collagen-vascular disease, and HIV infection. Pretreatment HRCT findings of ground-glass opacity (p = 0.004), a centrilobular pattern of ground-glass opacities (p = 0.003), interlobular septal thickening (p = 0.04), pericardial effusion (p = 0.04), pleural effusion (p = 0.01), and mediastinal lymphadenopathy (p = 0.009) were all more common in group 1, and strongly correlated with a risk of clinical worsening with epoprostenol treatment (Table 22-6, Fig. 22-12) (92).
TABLE 22-6 Frequency of HRCT Findings in Patients with Pulmonary Arterial Hypertension Who Had Epoprostenol Failure or Response
Pulmonary Hypertension Associated with Lung Disease or Hypoxemia
In patients who have PH resulting from lung disease, a pulmonary abnormality should be easily visible on HRCT (Fig. 22-13). Common lung diseases resulting in PH include emphysema, pulmonary fibrosis related to idiopathic pulmonary fibrosis or other fibrotic lung diseases, and COPD (68,93). The absence of a recognizable lung abnormality on HRCT in a patient who has known PH should suggest other causes than lung disease. Elevation of PAP as a result of lung disease is usually modest (68).
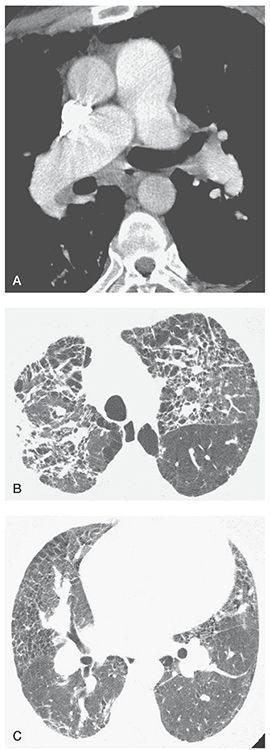
FIGURE 22-13 A–C: PH associated with end-stage sarcoidosis. A: On a contrast-enhanced spiral CT, enlargement of the main pulmonary artery to a diameter greater than that of the aorta is visible, as is dilatation of the right pulmonary artery. B: Extensive lung disease with pulmonary fibrosis is visible at a lung window setting. C: At a lower level, dilatation of the hilar pulmonary arteries is associated with findings of lung disease.
Although HRCT can be valuable in detecting lung disease in patients with known PH, it has been shown that HRCT may be of limited value in predicting the presence of PH in patients with lung fibrosis or interstitial lung disease (ILD) (94–97
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


