Chapter 19 Principles of Clinical Trials
Introduction
In items 1 and 2, the intervention, niacin use, is not under the control of the investigators. Clinical factors such as age, smoking status, and other comorbidities as well as HDL levels may affect the physician’s choice to prescribe niacin, so the statement that can be made on the basis of these studies is that niacin use is associated with changes in HDL. In contrast, when the intervention is randomized, factors that could be related to HDL and clinical characteristics that influence the decision to prescribe niacin are ignored. As a result, the statement that niacin use causes changes in HDL is valid. The causal relationship between a test treatment and an outcome is called efficacy when it is measured in clinical trials and effectiveness when it is measured in a routine care setting. RCTs are the gold standard for evidence of the efficacy of a drug or other intervention, and this evidence is used by the U.S. Food and Drug Administration (FDA) to determine whether new medical treatments should be approved.1
Hypothesis Testing
Niacin use raises HDL by at least 10% on average
Niacin use raises HDL by less than 10% or lowers HDL on average
Types of Comparisons
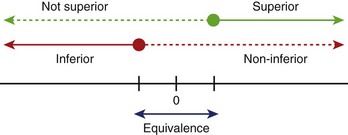
Hypotheses usually involve demonstrating that one treatment is significantly better than another (superiority) or that one treatment is not meaningfully worse than another treatment (non-inferiority, equivalence) (Figure 19-1). When testing an active drug or treatment compared with placebo, the study should be designed to detect the smallest clinically meaningful improvement as well as the cost and side effects of the drug. Placebo-controlled trials should always be superiority trials if the goal is to test the efficacy of the new drug. Sometimes non-inferiority placebo-controlled trials are used to establish the safety of a new drug.
Primary Outcome
The human phase of development for a drug or intervention is divided into four parts, and each part has unique objectives (Table 19-1). The choice of the outcome measure should be based on the goals of the development phase.
Table 19-1 Human Phase of Development for a Drug or Intervention
| PHASE | GOALS | EXAMPLES OF OUTCOMES |
|---|---|---|
| I | Maximum tolerated dose, toxicity, and safety profile | Pharmacokinetic parameters, adverse events |
| II | Evidence of biological activity | Surrogate markers* such as CRP, blood pressure, cholesterol, arterial plaque measure by intravascular ultrasound, hospitalization for CHF |
| III | Evidence of impact on hard clinical endpoints | Time to death, myocardial infarction, stroke, hospitalization for CHF, some combination of these events |
| IV | Postmarketing studies collecting additional information from widespread use of drugs or devices | Death, myocardial infarction, lead fracture, stroke, heart failure, liver failure |
CRP, C-reactive protein; CHF, congestive heart failure.
* A surrogate marker is an event that precedes the clinical event, which is (ideally) in the causal pathway to the clinical event. For example, if a drug is thought to decrease risk of myocardial infarction (MI) by reducing arterial calcification, changes in arterial calcification might be used as a surrogate endpoint because these changes should occur sooner than MI, leading to a reduction in trial time. Hospitalization is a challenging surrogate marker because it is not clearly in the causal pathway; hospitalization does not cause MI, but heart failure worsening to the degree that hospitalization is required is in the causal pathway. When hospitalization is used as a measure of new or worsening disease, it is important that the change in disease status and not just the hospitalization event is captured.
Randomization
Why Randomize?
Randomization, then, tends to even out any differences between the study participants assigned to one treatment arm compared with those to the other. The Coronary Artery Surgery Study (CASS) randomly assigned patients with stable class III angina to initial treatment with bypass surgery or medical therapy.2 If this trial had not been randomized and treatment assignment had been left to the discretion of the enrolling physician, it is likely that these physicians would have selected patients who they believed would be “good surgical candidates” for the bypass surgery arm. This would have led to a comparison of patients who were good surgical candidates receiving bypass surgery with a group of patients who, for one reason or another, were not good surgical candidates receiving medical treatment. It is likely that the medically selected patients would have been sicker, with more comorbidities than the patient selected for surgery. This design would not result in a fair comparison of the two treatment strategies. Randomization levels the playing field.
Intention to Treat
To make randomization work, analysis of RCT data needs to adhere to the principle of “intention to treat.” In its purest form, this means that data from each study participant are analyzed according to the treatment arm to which they were randomized, period. In the case of the Antiarrhythmics Versus Implantable Defibrillators (AVID) trial, this meant that data from patients randomly assigned to the implantable defibrillator arm were analyzed in that arm, whether or not they ever received the device.3 Data from patients randomly assigned to the antiarrhythmic drug treatment arm were analyzed with that arm, even if they had received a defibrillator. This may not make obvious sense, and many trial sponsors have argued that their new treatment just could not show an effect if the patient never received the new treatment. However, the principle of “intention to treat” protects the integrity of the trial by removing a large source of potential bias. In a trial to examine the efficacy of a new antiarrhythmic drug for preventing sudden cardiac death (SCD), for example, a sponsor might be tempted to count events only while the patient was still taking the drug. How, they would argue, could the drug have a benefit if the patient was not taking it? But, if, as has happened, the drug exacerbates congestive heart failure (CHF), then patients assigned to the experimental drug would be likely to discontinue taking the drug. And any subsequent SCD or cardiac arrests would not be attributed to the drug. In fact, it could be argued that the drugs led to a situation where the patient was more likely to die of an arrhythmia.
Stratification and Site Effect
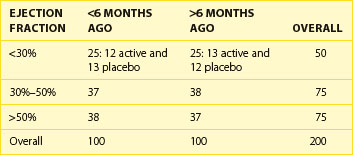
Some investigators get carried away with the concept of stratification to try to design randomization in such a way that all possible factors are balanced. It is easy to design a trial with too many strata. Take, for example, a trial stratifying on the basis of ejection fraction at baseline (<30%, 30% to 50%, and >50%) and a required history of myocardial infarction (MI) being “recent,” that is, within the past 6 months, or distant, that is, more than 6 months ago. With this, six strata have been created, a reasonable number for a sample size of, say, 200 or more subjects, randomized to one of two treatment options (Table 19-2). But if the decision is made to stratify by site, with 10 sites, for example, more strata than expected patients would be created. It has been shown that as the number of strata in a conventional randomization design is increased, the probability of imbalances between treatment groups is, in fact, increased as well.4
Types of Randomization Designs
Permuted block designs can lead to the same problems as with too many strata. For this reason, adaptive randomization is sometimes used. Several types of adaptive designs exist. Basically, the next randomization assignment is dependent, in some way, on the characteristics of the patients who have been randomized so far. Baseline adaptive techniques adjust the probabilities of assigning the next patient to one treatment arm or the other on the basis of the baseline characteristics of the patient compared with other patients already randomized.5 In the study design described above where randomization will be stratified by the class of angina, recent or distant history of MI, and ejection fraction, the objective is to keep a balance of treatment assignments within each stratum. So, as each patient is randomized, the randomization algorithm will look at the existing balance in that stratum and assign treatment on the basis of a biased coin toss.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


