Chapter 59 Arrhythmogenic Right Ventricular Cardiomyopathy
Historical Summary
In 1977, Fontaine et al reported six cases of sustained ventricular tachycardia (VT) in patients with enlarged right ventricles and hypothesized that this was a specific cardiomyopathy limited to the right ventricle.1 In 1982, Marcus et al coined the term arrhythmogenic right ventricular dysplasia (ARVD) in a report of 24 cases characterized by left bundle branch block (LBBB) pattern VT, wall motion abnormalities of the right ventricle (RV), and replacement of the RV myocardium by adipose and fibrous tissue.2 In 1994, the first diagnostic criteria were proposed. These were based on the identification of structural abnormalities, fibro-fatty replacement of the RV myocardium, particular electrocardiographic changes, arrhythmias of RV origin, and familial disease.3 This led to ARVC being recognized as a separate entity in the reclassification of cardiomyopathies by the World Health Organization/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyopathies.4–6 Over the following decade, various groups mapped familial forms of ARVC to a number of genetic loci.7–14 Molecular studies in a recessive, syndromic variant of ARVC, known as Naxos disease, led to identification of the first disease-causing mutation in the gene encoding plakoglobin, a major constituent of cell-to-cell junctions.15 This paved the way for the discovery of disease-causing mutations in other genes encoding desmosomal proteins in the more common autosomal dominant forms of ARVC.
Epidemiology
The prevalence of ARVC in the general population is difficult to determine because of the challenging nature of the diagnosis.16 Various studies in Europe reported a prevalence of between 0.6 and 4.4 per 1000, but it is unclear whether these represent local interest and expertise in the disease. ARVC is reported as a cause of SCD in 11% to 27% of individuals 35 years old and younger.17–19 The prevalence in one Italian cohort was found to be higher in athletes (22.4%) compared with nonathletes (8.2%).17
Pathology
In the original descriptions of ARVC, the RV cavity was described as typically dilated, and the RV wall was characterized by dome-shaped aneurysms, occurring particularly on the anterior surface of the pulmonary infundibulum, at the RV apex, and on the inferior wall of the RV (subsequently called the triangle of dysplasia). These macroscopic changes were accompanied by abundant subepicardial fat and endocardial thickening (Figure 59-1). Microscopically, a marked decrease was seen in myocardial fibers, with replacement of the myocardium with fatty tissue and fibrosis (Figure 59-2).2 Later, pathologic descriptions divided ARVC into two morphologic patterns: the fatty and the fibro-fatty forms, but fatty infiltration alone can be a normal phenomenon, particularly in older women.20
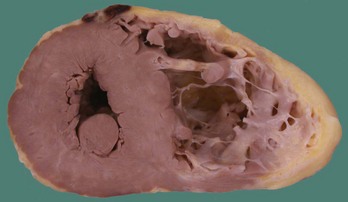
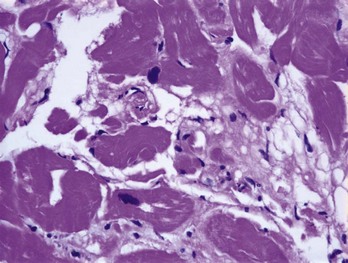
FIGURE 59-2 Histologic specimen of the right ventricle showing fatty infiltration and fibrosis within the myocardium.
(Courtesy Dr. Antonis Pantazis, Consultant Cardiologist, University College London Hospitals, UK.)
In a small series of studies on deaths due to ARVC, apoptosis was reported in 75% of patients.21 These apoptotic myocardial cells were frequently found in the myocardium not affected by fibro-fatty replacement, which suggests that this could account for the progressive loss of myocardial cells in the disease.22 Endomyocardial biopsy in 20 patients with ARVC revealed apoptosis in 35%. This was significantly associated with acute symptoms and signs and with a shorter (<6 months) clinical history, indicating that apoptosis is present in an early symptomatic stage of the disease.23
A number of postmortem reports described inflammatory infiltrates, and endomyocardial biopsies from some ARVC cases have been found to contain enteroviral ribonucleic acid (RNA) with homology to Coxsackie virus type B.21,24–26 In one study, the frequency of viral deoxyribonucleic acid (DNA) was similar to that observed in patients with myocarditis or dilated cardiomyopathy (DCM), but subsequent reports have failed to confirm these findings, perhaps reflecting patient selection and differences among inherited, sporadic, and nonfamilial forms of ARVC.27
Left Ventricular Involvement
By definition, the right ventricle must be affected to make a clinical diagnosis of ARVC, but it is well recognized that left ventricular involvement is common in patients fulfilling current diagnostic criteria. Pathologic examination of heart specimens at autopsy or cardiac transplantation revealed macroscopic and microscopic evidence of left ventricular involvement in up to 76% of cases.28 More recently, a study of 200 patients with ARVC undergoing cardiac magnetic resonance imaging (MRI) reported biventricular involvement in 56% of cases and predominant left-sided disease in 5%.29
Genetics
Systematic family studies have shown that ARVC is inherited in up to 50% of cases. Numerous genetic loci have been identified in linkage studies (Table 59-1). The mode of transmission is usually autosomal dominant, with a male predominance of 3 : 1 and variable penetrance, but autosomal recessive forms are well recognized and have provided the first insights into the genetic basis of ARVC.16
Table 59-1 Chromosomal Location, Involved Genes, and Mutations in Arrhythmogenic Right Ventricular Cardiomyopathy
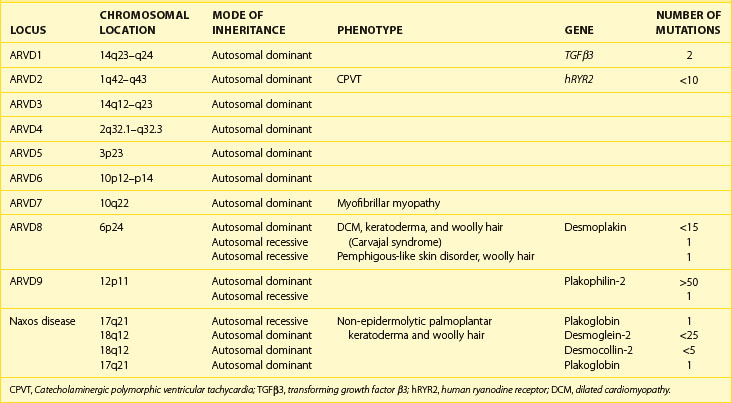
The first disease causing mutation was identified in an autosomal recessive syndromic form of ARVC, characterized by diffuse palmo-plantar keratoderma, woolly hair, and cardiomyopathy (Naxos disease).30 A homozygous mutation was identified in the gene encoding plakoglobin, a member of the armadillo protein family found in the adherens and desmosomal junctions between cardiac myocytes.15 Mutations in another desmosomal protein, desmoplakin, were subsequently identified in similar autosomal recessive syndromic forms of ARVC in South American and Arab families.31,32 Since then, heterozygous mutations in genes encoding these and other desmosomal proteins have been identified in patients with nonsyndromic autosomal dominant forms of ARVC.
The Intercellular Junction
Intercalated discs are specialized structures that provide mechanical and electrical coupling between adjacent cardiomyocytes. The intercalated disc is made up of three distinct structures: (1) gap junctions, (2) adherens junctions, and (3) desmosomes. The gap junction mediates the transfer of ions between cells, whereas the adherens junction (composed of cadherins, β-catenin, and γ-catenin [plakoglobin]) mediates the transmission of force between cells. The desmosome also mediates mechanical attachment between cells by linking the desmosomal cadherins, desmocollin, and desmoglein with the intermediate filament cytoskeleton.33,34 The intracellular components of the desmosomal cadherins interact with plakoglobin and plakophilin, which, in turn, bind to the N-terminal domain of desmoplakin, a plakin protein. The C-terminal of desmoplakin anchors desmin intermediate filaments to the cell surface.35 Another protein of the plakin family, plectin, is also present in desmosomes and contributes to the mechanical strength of cells.36,37 As well as providing cells with mechanical strength, the desmosome also plays an important role in tissue morphogenesis and differentiation.38 A simplified schematic representation of the organization of the cardiac desmosome is shown in Figure 59-3.
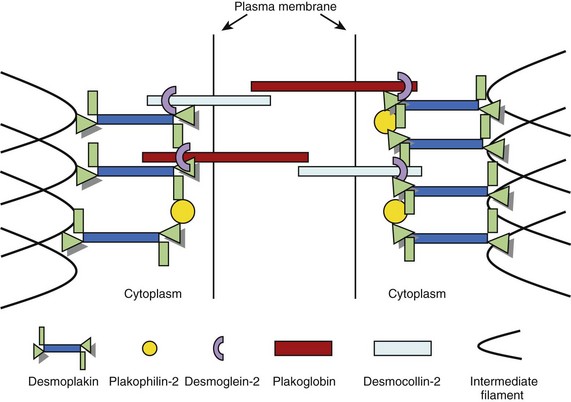
FIGURE 59-3 A schematic representation of the molecular desmosome.
(Data adapted from Green KJ, Gaudry CA: Are desmosomes more than tethers for intermediate filaments? Nat Rev Mol Cell Biol 1:208–216, 2000.)
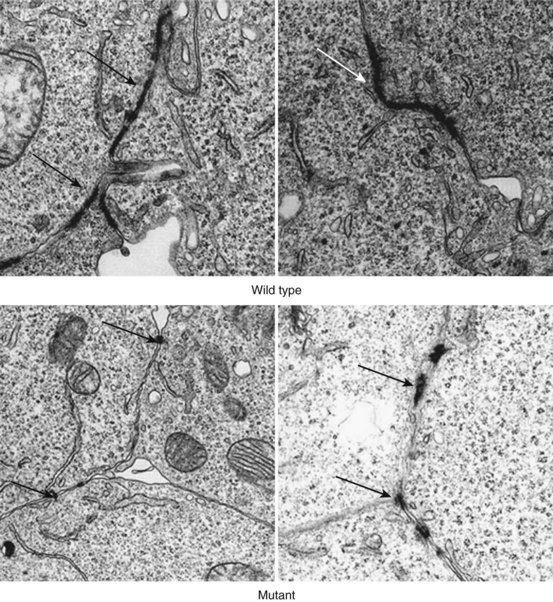
In 2002, the first mutation in the autosomal dominant form of ARVC was reported. This was found in the plakoglobin-binding domain of desmoplakin, and mutations in this gene have been reported in 6% to 16% of probands.14,39,40 Since then, heterozygous mutations have also been reported in plakoglobin, plakophilin-2 (PKP2), desmocollin-2 (DSC2), and desmoglein-2 (DSG2). PKP2 mutations are the most frequently found in patients with ARVC, with a reported prevalence between 27% and 43% of patents fulfilling diagnostic criteria.41 Over 50 individual mutations have been identified to date, but studies have not as yet revealed consistent genotype-phenotype correlations. The mechanism by which mutations result in disease is also unclear. It is suggested that mutations increase the susceptibility of the myocardium to the damaging effects of mechanical stress, thereby predisposing to cardiomyocyte detachment, death, and eventual replacement by fibro-fatty tissue.42 The predilection for the right ventricle has been explained by its thin wall and greater distensibility, but this is unlikely to be the sole explanation for disease in either ventricle.43 As desmosomal proteins interact with many other proteins, including key components of the cellular cytoskeleton and intermediate filaments, it is possible that ventricular dysfunction occurs as the result of reduced cytoskeletal integrity and impaired force transduction.37 Some desmosomal proteins (in particular plakoglobin) are also important signaling molecules that regulate the transcription of many other genes.44 Finally, a reduction in the number and size of gap junctions may result in an electrical coupling defect, thus increasing the propensity to arrhythmia, and could explain the occurrence of sudden death in patients without significant morphologic changes Figure 59-4.42
Nondesmosomal ARVC
Two nondesmosomal genes have been linked to ARVC: (1) the cardiac ryanodine receptor (hRYR2) and (2) transforming growth factor β3 (TGFβ3). Mutations in hRYR2 are more typically associated with effort-induced polymorphic VT and juvenile sudden cardiac death, without resting electrocardiogram (ECG) abnormalities or structural abnormalities, and mutations in this gene are no longer classified as a subtype of ARVC.45,46 Mutations in the intronic region of TGFβ3, which stimulates the production of extracellular matrix components, have been found to promoting myocardial fibrosis in vivo.47–49 Two ARVD1 kindred, however, lacked mutations in TGFβ3, which raises a question about its involvement in the pathogenesis of ARVC.50 Recently, a mutation in the transmembrane protein 43 (TNEM43) has been described (ARVD5). TNEM43 is predicted to be a cytoplasmic membrane protein without a cadherin domain, and the TNEM43 gene contains a response element for an adipogenic transcription factor.51
Phenocopies
When the left ventricle is involved and severe biventricular dysfunction is present, the differentiation from DCM can be difficult.52 Cardiac sarcoidosis can also mimic some of the clinical and structural abnormalities of ARVC but can be differentiated on endomyocardial biopsy.53 Right ventricular outflow tract VT can be difficult to distinguish from ARVC but generally has a good prognosis and can be successfully treated with catheter ablation therapy.54 Right ventricular myocarditis can mimic ARVC, and significant overlap between these two conditions exists, as inflammatory infiltrates are a feature of both.55,56 The histopathologic features of the heart in X-linked Emery-Dreifuss muscular dystrophy (EDMD) can mirror those of ARVC, but the two can be distinguished by family history and the presence of skeletal muscle involvement in EDMD.57
Symptoms and Physical Signs
Patients can present at any age with syncope, presyncope, or palpitations, In one case series, approximately one third of live probands gave a history of syncope, with over half reporting palpitations and presyncope.58 Other symptoms include chest pain or dyspnea often precipitated by exercise.59 In approximately 50% of cases, the first manifestation of ARVC is SCD, but in living patients, aborted SCD is the first manifestation in less than 10% of cases.28,60 Heart failure as a first presentation is probably uncommon; it accounted for less than 10% of cases in one series.58 Increasingly, patients are identified through family screening and are often asymptomatic. Cardiac examination in symptomatic and asymptomatic individuals is usually unremarkable.
Diagnosis
Because of the nonspecific nature of the clinical findings and the absence of a single diagnostic test, the diagnosis of ARVC is challenging. Recognition of these difficulties resulted in the formation of an International Task Force, which proposed standardized criteria for the diagnosis of ARVC in 1994.3 These criteria are based on the identification of structural, histologic, ECG, arrhythmic, and familial features, which are subdivided into major and minor criteria according to specificity. The presence of two major criteria, one major criterion plus two minor criteria, or four minor criteria from different categories is considered diagnostic (Table 59-2).3
Table 59-2 Task Force Criteria for the Diagnosis of Right Ventricular Dysplasia/Cardiomyopathy
| MAJOR CRITERIA | MINOR | |
|---|---|---|
| Global and/or regional dysfunction and structural alterations | Severe dilation and reduction of RV ejection fraction Localized RV aneurysms (akinetic or dyskinetic areas with diastolic bulging) Severe segmental dilation of the RV | Mild global RV dilation and/or ejection fraction reduction with normal LV Mild segmental dilation of the RV Regional RV hypokinesia |
| Tissue characterization of wall | Fibro-fatty replacement of myocardium on endomyocardial biopsy | |
| Repolarization abnormalities | Inverted T waves in right precordial leads (V2 and V3) in people aged >12 years, in absence of RBBB | |
| Depolarization/conduction abnormalities | Epsilon waves or localized prolongation (>110 ms) of QRS complex in right precordial leads (V1–V3) | Late potentials (SAECG) |
| Arrhythmias | LBBB-type ventricular tachycardia (sustained and non-sustained) by ECG, Holter, or exercise testing Frequent ventricular extrasystoles (>1000/24 hours on Holter) | |
| Family history | Familial disease confirmed at necropsy or surgery | Family history of premature sudden death (<35 years) from suspected RV dysplasia Familial history (clinical diagnosis based on present criteria) |
RV, Right ventricle; LV, left ventricle; RBBB, right bundle branch block; SAECG, signal-averaged electrocardiogram.
From McKenna WJ, Thiene G, Nava A, et al: Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy, Br Heart J 71:215–218, 1994.
The main goal of these guidelines was to establish a definitive diagnosis in probands, but tertiary referral center experience suggests that while the criteria are highly specific, they lack sensitivity for early disease.59 This led to the proposal of modified familial criteria for the diagnosis of ARVC in relatives of index cases. According to these criteria, the presence of a single disease feature (right precordial T-wave inversion, late potentials on signal averaged ECG, VT with LBBB morphology, or minor functional or structural abnormalities of the RV on imaging) in a relative of an index case should be considered diagnostic of ARVC (Table 59-3).58
Table 59-3 Familial Criteria for the Diagnosis of Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy
| Meet the familial criteria for a diagnosis of ARVC if ARVC is present in a first-degree relative plus one of the following: | |
| ECG | T-wave inversion in right precordial leads (V2 and V3) |
| SAECG | Late potentials seen on signal-averaged ECG |
| Arrhythmia | LBBB-type VT on ECG, Holter or during exercise testing Extrasystoles >200 over 24-hour period |
| Structural or functional abnormalities of the RV | Mild global RV dilation or EF reduction with normal LV Mild segmental dilation of the RV Regional RV hypokinesia |
ARVC, Arrhythmogenic right ventricular cardiomyopathy; ECG, electrocardiogram; SAECG, signal-averaged electrocardiogram; LBBB, left bundle branch block; VT, ventricular tachycardia; RV, right ventricle; EF, ejection fraction.
Modified from Hamid MS, Norman M, Quaraishi A, et al: Prospective evaluation of relatives for familial arrhythmogenic right ventricular dysplasia/cardiomyopathy reveals a need to broaden diagnostic criteria, J Am Coll Cardiol 40(8):1445–1450, 2002.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


