Pathophysiology of Hypertension: Introduction
More than 1 billion individuals worldwide, including 50 million Americans, have high blood pressure warranting some form of treatment.1-4 Higher-than-optimal blood pressure is the number one attributable risk for death throughout the world, and approximately 7.6 million deaths per year are attributed to uncontrolled hypertension.3 As life expectancy continues to increase, hypertension will become an even more important medical and public health issue because blood pressure typically increases with aging in most industrialized countries. In the United States, 50% of people 60 to 69 years old and approximately 75% of people 70 years old and older have hypertension.1,2 In some isolated, nonindustrialized populations, however, blood pressure does not increase with increasing age, and only a small fraction of the population develops hypertension. This suggests that predisposing environmental factors play a major role in causing hypertension and that an increase in blood pressure with aging is not inevitable when these factors are absent.
A direct positive relationship between blood pressure and cardiovascular disease (CVD) risk has been observed in men and women of all ages, races, ethnic groups, and countries, regardless of other risk factors for CVD.4 Observational studies indicate that death from CVD increases progressively and linearly as blood pressure increases above 115 mm Hg systolic and 75 mm Hg diastolic pressure.3 For every 20-mm Hg systolic or 10-mm Hg diastolic increase in blood pressure, there is a doubling of mortality from both ischemic heart disease and stroke in all age groups from 40 to 89 years old.5
Classification of Hypertension
Blood pressure is a variable quantitative trait with a normal distribution that is slightly skewed to the right. Although there is no clear level of blood pressure where CVD begins to occur, a definition of hypertension, although somewhat arbitrary, is useful for making decisions about treatment. A commonly used blood pressure classification was proposed in 2003 by the Seventh Report of the United States Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC 7) (Table 69–1).6 This classification is based on the average of two or more blood pressure readings after an initial screening visit and is for individuals who are not taking antihypertensive medication and who are not acutely ill. When systolic and diastolic blood pressures fall into different categories, the JNC 7 recommends that the higher category be selected to classify the person’s blood pressure.
| Category | Systolic Blood Pressure (mm Hg) | Diastolic Blood Pressure (mm Hg) | |
|---|---|---|---|
| Normalb | <120 | and | <80 |
| Prehypertension | 120-139 | and | 81-89 |
| Hypertensionc | |||
| Stage 1 | 140-159 | or | 90-99 |
| Stage 2 | ≤60 | or | ≤100 |
According to JNC 7 criteria, normal blood pressure is defined as a systolic blood pressure below 120 mm Hg and a diastolic blood pressure below 80 mm Hg. Persons with a systolic blood pressure between 120 and 139 mm Hg or diastolic blood pressure between 80 and 89 mm Hg are designated as having prehypertension. The diagnosis of hypertension is made by a confirmed systolic blood pressure of 140 mm Hg or above or a diastolic blood pressure of 90 mm Hg or above. Hypertension is further characterized into two stages according to the patient’s level of systolic and diastolic blood pressure. Stage 1, the milder (systolic, 140-159 mm Hg or diastolic, 90-99 mm Hg) and most common form of hypertension, accounts for approximately 80% of hypertension. Stage 2 hypertension includes those with systolic blood pressure of 160 mm Hg or above or diastolic blood pressure of 100 mm Hg or above. Isolated systolic hypertension is defined as systolic blood pressure of 140 mm Hg or above and diastolic blood pressure below 90 mm Hg and staged appropriately.
Using these definitions and including those who are taking antihypertensive medication, approximately 24% of the adult population in the United States has hypertension.6 This percentage varies with (1) race, being higher in blacks (32%) and lower in whites (23%) and Mexican Americans (23%); (2) age because systolic blood pressure increases throughout life in the United States, as well as in most industrialized countries, but diastolic blood pressure increases until age 55 to 60 years; (3) gender, with hypertension being more prevalent in men than in premenopausal women; after menopause, however, women have blood pressures that are nearly the same as in men; (4) geographic patterns, with hypertension being more prevalent in the southeastern United States; and (5) socioeconomic status, which is inversely related to the prevalence, morbidity, and mortality rates of hypertension.
Definition of Primary (Essential) Hypertension
Primary (essential) hypertension, which accounts for 95% of all cases of hypertension, has been traditionally defined as high blood pressure for which an obvious secondary cause (eg, renovascular disease, aldosteronism, pheochromocytoma, or gene mutations) cannot be determined.
Although primary hypertension is a heterogeneous disorder, some of the main causes of high blood pressure in primary hypertension are known. For example, overweight and obesity may account for as much as 65% to 75% of the risk for primary hypertension, as discussed later in this chapter. Other factors, such as sedentary lifestyle, excess intake of alcohol or salt, and low potassium intake, are also known to increase blood pressure in many patients who are classified as having primary hypertension. Therefore, it is probably inappropriate to define primary hypertension as an increase in blood pressure without cause because identified causes can be found in many patients.7
This chapter discusses basic concepts of circulatory regulation, the physiologic mechanisms involved in short- and long-term control of blood pressure, and the pathophysiologic changes in the major blood pressure control systems that can lead to a few types of secondary hypertension, as well as primary hypertension. We also review interactions between genetic and environmental factors that influence intermediate phenotypes such as activity of the sympathetic nervous system (SNS), the renin–angiotensin system (RAS), endothelial factors, oxidative stress, and natriuretic hormones, which, in turn, influence vascular resistance, cardiac output, renal excretion of salt and water, and therefore blood pressure.
Basic Concepts of Circulatory Control
A discussion of cardiac output regulation often begins with the well-known formula Cardiac output = Stroke volume × Heart rate. This equation provides a conceptual framework that is adequate to describe the general determinants of cardiac pumping, but it does not elucidate another major determinant of cardiac output—the venous return (total tissue blood flow). To illustrate this point, consider the following question: Which of the following changes in cardiac output would you expect to find 7 days after surgical reduction of kidney mass by 50% (removal of one kidney): an increase, a decrease, or no change? The correct answer is a decrease in cardiac output. Why is cardiac output reduced by removal of a kidney when there has been no obvious effect on cardiac pumping ability or heart rate? This is not easy to comprehend if we think in a “cardiocentric” manner. However, the answer becomes obvious if we consider that cardiac output is, in the steady state, equal to the sum of the blood flows of all of the tissues.
Figure 69–1 illustrates the important relationship between cardiac output and tissue blood flow and shows why removing a kidney or any of other organ reduces cardiac output. Removing one kidney decreases venous return to the heart by approximately 10% (assuming that total flow to both kidneys is approximately 20% of the cardiac output). There is very little change in blood volume because the remaining kidney is able to rapidly increase its excretion to match intake of water and electrolytes. A reduction in cardiac output would also be observed with amputation of an arm or a leg or removal of any other tissue from the body. Conversely, an increase in cardiac output would occur when additional tissue is added to the body such as occurs with normal growth or excess weight gain. These examples illustrates that cardiac output is determined not only by the function of the heart but also by the peripheral circulation. Except when the heart is severely weakened and unable to adequately pump the venous return, cardiac output (total tissue blood flow) is determined mainly by the metabolic needs and other special needs of the tissues and organs of the body, although intrinsic and neurohumoral mechanisms allow the heart to effectively accommodate changes in venous return.

This conceptual framework is helpful in explaining changes in cardiac output during exercise (when metabolic activity and blood flow to skeletal muscles are increased), after eating a large meal (which increases metabolic activity and blood flow in the gastrointestinal system), and in pathophysiologic conditions such as hypertension in which cardiac output is also determined largely by the metabolic demands of the tissues. In most circumstances, cardiac pumping ability plays mainly a permissive role in determining the cardiac output.
The importance of integrating the principles of tissue blood flow regulation in discussing cardiac output can be illustrated by consideration of the chronic effects of vasodilators and vasoconstrictors. In most cases, cardiac output changes very little even when there are high levels of circulating vasoconstrictors (eg, angiotensin II [Ang II]) or vasodilators (eg, calcium channel blockers). If cardiac output regulation is the sum of all local blood flow regulations, why is cardiac output not significantly altered in these conditions?
To answer this question, we must consider one of the most fundamental principles of circulatory function—the ability of each tissue to autoregulate its own blood flow according to the metabolic needs and other functions of the tissue.8-10 Administration of a powerful vasoconstrictor, such as Ang II, may cause a transient decrease in cardiac output but usually has little long-term effect if it does not alter metabolic rate of the tissues. Likewise, vasodilators cause only short-term changes in tissue blood flow and cardiac output if they do not alter tissue metabolism.
Therefore, to effectively explain cardiac output regulation, it is necessary to discuss the mechanisms that control blood flow in the different tissues. Local blood flow regulation, as is true for most physiologic control systems, involves short- and long-term mechanisms. Acute control occurs within seconds or minutes as a result of constriction or dilatation of the vasculature. After administration of a vasoconstrictor that does not alter metabolic rate of the tissues, there is a transient decrease in the supply of nutrients and oxygen to the tissues and an accumulation of metabolic waste products. This, in turn, causes vasodilation and a return of tissue blood flow toward normal. In some tissues where blood flow regulation is not determined mainly by metabolic needs, such as the kidney, some vasoconstrictors, such as Ang II, may cause a small, sustained decrease in blood flow that barely alters cardiac output. Other short-term controls, such as the myogenic response, also alter vascular resistance in response to changes in blood pressure and help to autoregulate blood flow in the different tissues.11
Long-term blood flow regulation takes place over several days or weeks and involves structural changes in the blood vessels, such as thickening of vessel walls and decreased numbers of capillaries (rarefaction) in some tissues when blood pressure is chronically elevated. When tissues grow, additional blood vessels are generated (angiogenesis or vasculogenesis) to provide the required blood flow and metabolic substrates for the tissues. Together, the short- and long-term mechanisms maintain the required levels of blood flow in each tissue to ensure normal tissue function. Thus, in most physiologic conditions, excluding those associated with impaired cardiac pumping ability, the cardiac output reflects mainly the combined actions of the multiple control mechanisms for blood flows in the different tissues of the body.
In conditions such as heart failure or when large increases in cardiac output are needed to meet the metabolic demands of the body’s tissues, such as exercise, the various factors that alter cardiac pumping also play a major role in regulating cardiac output.
The main function of the circulation is to provide adequate blood flow to each tissue to meet its requirements. This is achieved by a combination of local tissue controls that regulate vascular tone as well as overall adjustments of the circulation that influence cardiac pumping and vascular tone.10,12 For example, during intense exercise, local conditions (eg, accumulation of metabolites or decreased levels of oxygen and nutrients) in the skeletal muscles cause intense vasodilation that permits adequate blood flow to match the increased metabolic requirements of the muscles. However, this also reduces peripheral vascular resistance, which tends to decrease blood pressure. Normally, this is offset by multiple neurohumoral changes (eg, sympathetic stimulation) that tend to elevate the blood pressure.
If, for some reason, peripheral vasodilation continues for several days, such as occurs with anemia or with opening a large arteriovenous fistula, additional adjustments take place that cause salt and water retention by the kidneys and increased blood volume or even hypertrophy of the heart if the stimulus lasts for several weeks. Thus, the multiple factors that control the circulation, including those that influence cardiac output, blood pressure, blood volume, and others, normally work in concert to provide adequate blood flow to the tissues of the body.
These same control mechanisms also operate in hypertension. However, one of the important characteristics of many, but not all, hypertensive patients is that they have increased total peripheral vascular resistance (TPR). Does this also mean that blood flow to the tissues is impaired? Excluding those hypertensive persons with heart failure or severe target-organ injury, the answer to this question is generally no. In most instances, tissue blood flows are approximately the same in normotensive and hypertensive subjects and are regulated at a level that is adequate to supply the needs of the tissues.13 The elevated vascular resistance observed in many patients with hypertension appears to be an autoregulatory response that helps to maintain normal tissue blood flow despite the increased blood pressure rather than a primary cause of the hypertension. Thus, the hemodynamic pattern that is often (but not always) observed in nonobese subjects with essential hypertension is normal blood flow, normal oxygen consumption, and elevated vascular resistance.13
Cerebral blood flow, for example, shows a normal value of about 50 mL/min/100 g per tissue weight in essential hypertension. Coronary blood flow is elevated in essential hypertension in proportion to the increase in myocardial hypertrophy. Blood flow per unit weight of heart muscle is usually normal, however, with a value of about 80 mL/min/100 g per tissue weight. Splanchnic blood flow is slightly reduced in essential hypertension, having a typical value of about 750 mL/min/m2 of surface area compared with about 800 mL/min/m2 in normotensive subjects. Skin blood flow is also normal in individuals with essential hypertension.
Renal blood flow has been observed to be increased, normal, or decreased in those with essential hypertension.13,14 These seemingly disparate observations, however, should be interpreted with regard to the special functional needs of the kidney and the conditions under which renal blood flow is studied. For example, increased dietary protein, high sodium intake, and excess weight gain all are associated with increased renal blood flow. Nephron loss, which may occur with prolonged, uncontrolled hypertension and diabetes, leads to reduction in renal blood flow. Impaired renal blood flow is also related to the cause of hypertension in some individuals.
Although resting skeletal muscle blood flow is, for the most part, normal in those with essential hypertension, several differences in blood flow regulation have been noted. For example, the ability of skeletal muscle blood vessels to dilate in some patients with essential hypertension is impaired.15 This reduced blood flow “reserve” in hypertension is probably a result of structural limitations imposed by blood vessel hypertrophy and of endothelial dysfunction and impaired release of nitric oxide (NO).
In general, the maximal level of exercise as quantified by oxygen uptake is depressed in proportion to the severity of hypertension. Arterial pressure is high before exercising and increases even further during exercise. In the presence of impaired vasodilation, elevated blood pressure boosts blood flow through the skeletal muscle but it also increases cardiac afterload, which limits both cardiac output and exercise performance. At each level of exercise below maximum, however, cardiac output and skeletal muscle blood flow are generally identical to flows seen in normotensive subjects.13 These flows are achieved at higher vascular resistances and higher blood pressures; the resistance and blood pressure effects cancel each other to yield a normal blood flow in most tissues.
In obese hypertensive patients, resting skeletal muscle blood flow per gram of tissue weight may be somewhat elevated compared with lean individuals; however, the increase in skeletal muscle blood flow that occurs in exercise is attenuated and the forearm reactive hyperemia after temporary occlusion of the brachial artery patients is less in obese than in lean normotensive subjects.16 Thus, although obesity-associated hypertension may be associated with increased resting blood flow, flow reserve is often reduced.
In older hypertensive individuals, total tissue blood flow (ie, cardiac output) is often reduced compared with the flow in younger individuals. This is perhaps not surprising if one considers that lean muscle mass usually decreases with aging. Because cardiac output is a sum of all tissue blood flows, decreased muscle mass and decreased physical activity characteristic of older hypertensive patients also are associated with reduced cardiac output and decreased total body oxygen consumption.
Adequate tissue blood flow in response to normal daily activities requires an adequate blood pressure. This is illustrated by the response to physical exercise in patients with autonomic dysfunction. As a person with normal autonomic reflexes begins to exercise, skeletal muscle vascular resistance is reduced, but skeletal muscle blood flow and cardiac output increase markedly and blood pressure remains relatively constant. In persons with autonomic dysfunction, exercise also decreases skeletal muscle vascular resistance, but blood pressure decreases and muscle blood flow and cardiac output increase only modestly because of impaired autonomic reflexes. Therefore, exercise is not well tolerated, and syncope often occurs.
When cardiac output is inadequate to meet normal tissue needs, as occurs in heart failure or severe hypovolemia, strong activation of the SNS, the RAS, and other hormonal factors produce vasoconstriction that may override normal flow regulation in some organs, such as skeletal muscle and skin. This keeps blood pressure from falling too low and provides adequate blood flow to the vital organs, especially the brain and the heart.
The most important function of blood pressure is to provide the driving force that moves blood through the vascular system to supply the needs of the tissues. Consequently, the regulation of blood pressure is a complex physiologic function that depends on the integrated actions of multiple cardiovascular, renal, neural, endocrine, and local tissue control systems.
Blood pressure varies considerably throughout the day depending on the activity of the body, environmental influences, and the responses of these multiple blood pressure control systems. Hypertension is usually considered to be a disorder of the average level at which blood pressure is regulated, although there is increasing interest in other measures of blood pressure, including peak arterial pressure, lability of blood pressure, nighttime and daytime blood pressure, blood pressure responses to stress, and so forth. Many of the cardiovascular derangements associated with hypertension, such as cardiac and vascular hypertrophy, arise as compensatory mechanisms for the hypertension, and reducing blood pressure can largely reverse these changes if they have not progressed too far.
The multiple local control, hormonal, neural, and renal systems that regulate blood pressure are often discussed in terms of how they influence cardiac pumping or vascular resistance because of the well-known formula Mean arterial pressure = Cardiac output × Total peripheral resistance. This conceptual framework (with the addition of factors that influence vascular capacity and transcapillary exchange) is adequate to explain short-term blood pressure regulation but is inadequate when discussing abnormalities of long-term blood pressure regulation, such as hypertension.
To illustrate this point, let us consider the following question: What changes in blood pressure, cardiac output, and extracellular fluid volume would you expect to find several days after a 50% increase in TPR caused by closure of a large arteriovenous (AV) fistula? In this case, cardiac pumping ability is not directly altered and TPR is chronically increased by 50%. One might assume that blood pressure would also rise chronically if increased TPR is sustained. However, several days after closure of an AV fistula, there are no detectable changes in mean arterial pressure despite a sustained increase of TPR. Likewise, increasing TPR by amputation of a limb or hypothyroidism (which reduces metabolic rate of the tissues and increases vascular resistance) or decreasing TPR by creating an AV fistula, anemia, or hyperthyroidism fails to have a significant long-term effect on mean arterial pressure (Fig. 69–2). Why do these large chronic changes in TPR have no significant long-term effect on mean arterial pressure? To explain blood pressure regulation in these circumstances, we must introduce two other concepts: (1) time dependency of blood pressure control mechanisms and (2) the necessity of maintaining balance between intake and output of water and electrolytes and the role of blood pressure in maintaining this balance.
Figure 69–2
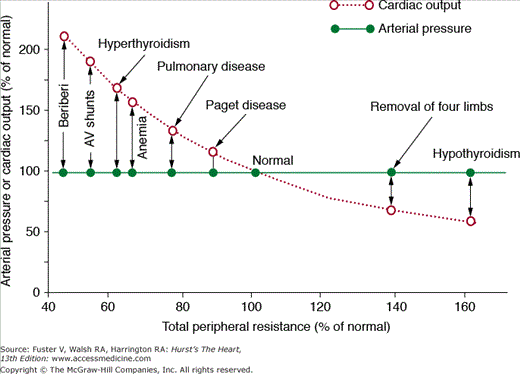
Failure of changes in total peripheral vascular resistance (TPR) to cause chronic changes in arterial pressure in several clinical conditions in which the kidneys are functioning normally. In each case, there is a reciprocal relationship between TPR and cardiac output but no long-term effect on arterial pressure. This figure was published in Textbook of Medical Physiology. 11th ed. Guyton AC, Hall JE. Copyright Elsevier (2006).
The effectiveness of the different feedback systems for blood pressure control can be expressed in terms of feedback gain, which is defined as the amount of “correction” by the control system divided by the remaining “error” after a perturbation. For example, if a disturbance initially increases blood pressure from 100 to 150 mm Hg and a control system returns blood pressure to 125 mm Hg, the correction would be −25 mm Hg, and the remaining error would be +25 mm Hg. Therefore the feedback gain would be −25/+25, or −1.0, indicating a negative feedback that corrects one half of the initial disturbance. The higher the feedback gain, the more powerful the control system.
Blood pressure control systems are often considered as if they were static, and time dependency is usually not discussed. Short-term control mechanisms are often emphasized to a greater degree than long-term controls, probably because they have been studied much more extensively and are easier to explain, even though most cardiovascular disorders, including hypertension, involve abnormalities of long-term regulation.
If we examine the maximal feedback gains of several blood pressure controllers, it is obvious that their quantitative importance is highly time dependent. Figure 69–3 shows the response of some of the major control systems after a sudden change in blood pressure, as might occur with rapid blood loss. Three important neural control systems begin to function powerfully within seconds: (1) the arterial baroreceptors, which detect changes in blood pressure and send appropriate autonomic reflex signals back to the heart and blood vessels to return the blood pressure toward normal; (2) the chemoreceptors, which detect changes in oxygen or carbon dioxide in the blood and initiate autonomic feedback responses that influence blood pressure; and (3) the central nervous system (CNS), which responds within a few seconds to ischemia of the vasomotor centers in the medulla, especially when the blood pressure falls below about 50 mm Hg. Each of these nervous control mechanisms works rapidly and can have potent effects on blood pressure. Also note, however, that the feedback gains of these systems decreases with time, as a disturbance of blood pressure is maintained.
Figure 69–3
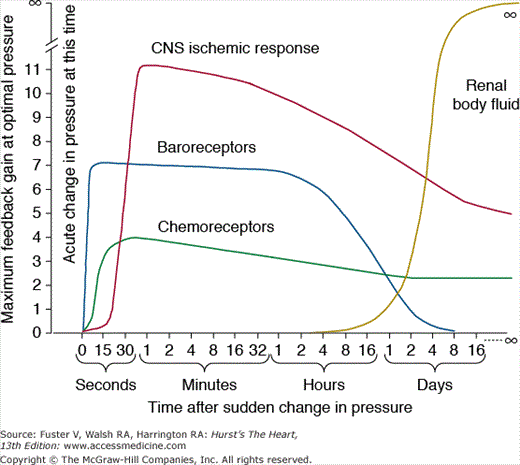
Time dependency of blood pressure control mechanisms. Approximate maximum feedback gains of various blood pressure control mechanisms at different time intervals after the onset of a disturbance to arterial pressure. CNS, central nervous system. This figure was published in Textbook of Medical Physiology. 11th ed. Guyton AC, Hall JE, 230, Copyright Elsevier (2006).
Within a few minutes or hours after a blood pressure disturbance, several additional control systems react, including (1) a shift of fluid from the interstitial spaces into the blood stream in response to decreased blood pressure (or a shift of fluid out of the blood into the interstitial spaces in response to increased blood pressure); (2) the RAS, which is activated when blood pressure falls too low and suppressed when blood pressure increases above normal; and (3) multiple vasodilators systems (not shown in the figure) that are suppressed when blood pressure decreases and stimulated when blood pressure increases above normal.
Most of the blood pressure regulators are proportional control systems. This means that they will correct a blood pressure abnormality only part of the way back toward the normal level but never all the way back. The arterial baroreceptor reflex system, for example, has a proportional feedback gain of approximately 2.0 during acute changes in blood pressure and therefore buffers about two-thirds of a sudden change in the blood pressure.10
There is one blood pressure control system, the renal–body fluid feedback system, with near infinite feedback gain if it is given enough time to operate.17,18 Thus, the renal–body fluid feedback control mechanism does not stop functioning until the arterial pressure returns nearly all the way back to its original control level, as discussed below.
Figure 69–4 shows the conceptual framework for understanding long-term control of blood pressure by the renal–body fluid feedback mechanism. Extracellular fluid volume is determined by the balance between intake and excretion of salt and water by the kidneys. Even a temporary imbalance between intake and output can lead to a change in extracellular volume and potentially a change in blood pressure. Under steady-state conditions, there must always be a precise balance between intake and output of salt and water; otherwise, there would be continued accumulation or loss of fluid leading to complete circulatory collapse within a few days. In fact, it is more critical to maintain salt and water balance than to maintain a normal level of blood pressure and, as discussed below, increased blood pressure is a means of regulating these balances in the face of impaired kidney function.
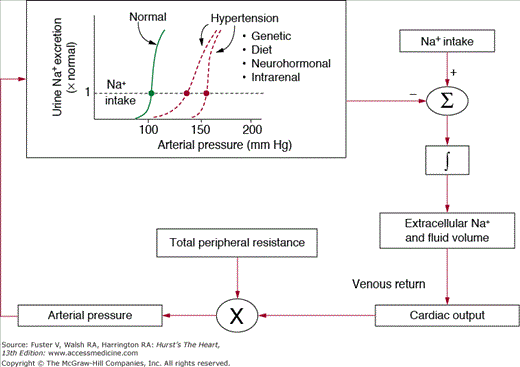
A key mechanism for regulating salt and water balance is pressure natriuresis and diuresis, the effect of increased blood pressure to raise sodium and water excretion.18,19 Under most conditions, this mechanism acts to stabilize blood pressure and body fluid volumes. For example, when blood pressure is increased above the renal setpoint, because of increased TPR or increased cardiac pumping ability, this also increases sodium and water excretion via pressure natriuresis. As long as fluid excretion exceeds fluid intake, extracellular fluid volume will continue to decrease, reducing venous return and cardiac output until blood pressure returns to normal and fluid balance is reestablished.
An important feature of pressure natriuresis is that various hormonal and neural control systems can amplify or blunt the basic effects of blood pressure on sodium and water excretion.19 For example, during chronic increases in sodium intake, only small changes in blood pressure are needed to maintain sodium balance in most people. One reason for this insensitivity of blood pressure to changes in salt intake is decreased formation of antinatriuretic hormones such as Ang II and aldosterone, which enhance the effectiveness of pressure natriuresis and allow sodium balance to be maintained with little or no increase in blood pressure. On the other hand, excessive activation of these antinatriuretic systems can reduce the effectiveness of pressure natriuresis, thereby necessitating greater increases in blood pressure to maintain sodium balance.
Another important feature of pressure natriuresis is that it continues to operate until blood pressure returns to nearly the original setpoint. In other words, it acts as part of a near infinite gain feedback control system.17 As far as we know, it is the only feedback system for blood pressure regulation in the body that displays near infinite feedback gain, and this property makes it a dominant long-term controller of blood pressure.
Figure 69–5 illustrates the near infinite gain characteristic of the renal–body fluid feedback system. In this case, blood pressure is increased without a change in pressure natriuresis, as would occur with increased TPR because of closure of an AV fistula, coarctation of the aorta below the kidneys (aortic coarctation above the kidneys causes marked hypertension), or other changes that increase peripheral vascular resistance without influencing renal vascular resistance. The peripheral constriction initially increases blood pressure from point A to point B, but the increase in blood pressure cannot be sustained; as long as pressure natriuresis is unaltered, sodium excretion will increase above intake, thereby reducing extracellular fluid volume and cardiac output until blood pressure eventually returns all the way back to normal. In fact, the normal blood pressure is the only point at which sodium and water balance can be maintained. Likewise, disturbances that decrease peripheral vascular resistance or alter cardiac function without influencing renal pressure natriuresis have no long-term effect on arterial pressure.17,18
Figure 69–5

Long-term effects of increased total peripheral resistance (TPR), such as that caused by closure of a large arteriovenous fistula, with no change in the renal pressure natriuresis relationship. Blood pressure is initially increased from point A to point B, but elevated blood pressure cannot be sustained because sodium excretion exceeds intake, reducing extracellular fluid volume until blood pressure returns to normal and sodium balance is reestablished. Redrawn from Hall.14
In all forms of human or experimental hypertension studied thus far, there is a shift of pressure natriuresis that appears to initiate and sustain the hypertension. In some cases, abnormal kidney function is caused by intrarenal disturbances that alter renal hemodynamics or increased tubular reabsorption. In other cases, the altered kidney function is caused by extrarenal disturbances, such as increased SNS activity or excessive formation of antinatriuretic hormones, that reduce the kidney’s ability to excrete sodium and water and eventually increase arterial pressure. Consequently, effective treatment of patients with hypertension requires interventions that reset pressure natriuresis toward normal levels of blood pressure either by directly increasing renal excretory capability (eg, with diuretics), or by reducing antinatriuretic influences (eg, with RAS blockers) on the kidneys.
Renal Mechanisms of Hypertension
For more than 100 years, severe kidney disease has been recognized as closely associated with hypertension. However, the importance of more subtle renal dysfunction in the pathogenesis of essential hypertension has not been widely appreciated, partly because there are no obvious renal defects in most patients with primary hypertension. Many of the measurements commonly used to evaluate kidney function, such as glomerular filtration rate (GFR), renal blood flow, serum creatinine, and sodium excretion are often within the normal range, at least in the early stages of hypertension before damage to the kidneys has occurred. On the other hand, increased TPR is an obvious abnormality found in most patients with hypertension, leading many to emphasize peripheral vasoconstriction as a cause of increased blood pressure. However, as discussed previously, increased peripheral vascular resistance appears to be an autoregulatory response to increased blood pressure in many hypertensive subjects and in the absence of altered kidney function is not a cause of sustained hypertension.
An observation that points toward abnormal kidney function as a key factor in hypertension is that almost all forms of experimental hypertension, as well as all monogenic forms of human hypertension thus far discovered, are caused by obvious insults to the kidneys that alter renal hemodynamics or tubular reabsorption. For example, constriction of the renal arteries (eg, Goldblatt hypertension), compression of the kidneys (eg, perinephritic hypertension), or administration of sodium-retaining hormones (eg, mineralocorticoids or Ang II) are all associated with either initial reductions in renal blood flow and GFR or increases in renal tubular reabsorption prior to development of hypertension. Likewise, in all known monogenic forms of human hypertension, the common pathway to hypertension appears to be increased renal tubular sodium reabsorption caused by mutations that directly increase renal electrolyte transport or the synthesis or activity of antinatriuretic hormones. As blood pressure increases, the initial renal changes are often obscured by compensations that restore kidney function toward normal. The increase in blood pressure then initiates a cascade of cardiovascular changes, including increased peripheral vascular resistance (which helps to maintain normal tissue blood flow) that may be more striking than the initial disturbance of kidney function. For this reason, the importance of renal dysfunction in causing hypertension has often been underestimated.
Although specific abnormalities of kidney function are difficult to identify in most patients with primary hypertension, the one aspect of kidney function that is abnormal in all types of experimental and clinical hypertension is renal pressure natriuresis.14,18 The fact that a normal rate of sodium excretion (equal to sodium intake) is maintained in chronic hypertension despite elevated blood pressure, which would normally cause natriuresis and diuresis, indicates that pressure natriuresis is reset in hypertensive subjects.
Because tubular reabsorption and GFR are both approximately 100-fold greater than urinary excretion, relatively small changes in either of these variables can have large effects on urinary excretion. Any long-term reduction in GFR, for example, must be perfectly compensated for by mechanisms that either restore GFR or decrease tubular reabsorption if renal excretion is to be returned toward normal and sodium and fluid balance are maintained. Likewise, any chronic increase in tubular reabsorption must be compensated for by increased GFR or by a restoration of tubular reabsorption to normal. Otherwise, fluid retention would continue eventually causing circulatory collapse.
If intrinsic renal mechanisms or neurohumoral adjustments are capable of returning renal excretion to normal in the face of insults to the kidneys, hypertension may not develop. However, the fact that hypertension occurs so frequently indicates that intrinsic renal mechanisms and neurohumoral controls may not be powerful enough to completely prevent alterations in renal excretion when there are major abnormalities of GFR or tubular reabsorption. In these instances, increased arterial pressure helps to maintain glomerulotubular balance and normal rates of sodium and water excretion, equal to intake, despite abnormal kidney function. The general types of renal abnormalities that can cause chronic hypertension include increased preglomerular resistance, decreased glomerular capillary filtration coefficient, reduced numbers of functional nephrons, and increased tubular reabsorption (Table 69–2 and Fig. 69–6).
| Causes | Blood Pressure | Pressure Natriuresis | Renal Blood Flow | Glomerular Filtration Rate | Plasma Renin Activity | Glomerulosclerosisb |
|---|---|---|---|---|---|---|
| Salt-sensitive hypertension | ||||||
| 1. Decreased kidney mass | ↑, ↔ | Decreased slope | ↓ | ↓ | ↓ | Yes |
| 2. Decreased glomerular capillary filtration coefficient (Kf) | ↑ | Decreased slope | ↑ | ↓, ↔ | ↑, ↔ | Yes |
| 3. Increased distal and collecting tubule reabsorption | ↑ | Decreased slope | ↑ | ↑ | ↓ | Yes |
| Salt-insensitive hypertension | ||||||
| 1. Increased preglomerular resistance | ↑ | Parallel shift | ↓, ↔ | ↓, ↔ | ↑, ↔ | No |
| 2. Increased reabsorption in proximal tubules and loops of Henle | ↑ | Parallel shift | ↑ | ↑ | ↑, ↔ | Yes |
Figure 69–6

Steady-state relationships among arterial pressure and urinary sodium excretion and sodium intake for subjects with normal kidneys and four general types of renal dysfunction that cause hypertension: decreased kidney mass, increased reabsorption in distal and collecting tubules, reductions in glomerular capillary filtration coefficient (Kf), and increased preglomerular resistance. Note that whereas increased preglomerular resistance causes salt-insensitive hypertension, the other renal abnormalities cause salt-sensitive hypertension.
Examples of a generalized increase in preglomerular resistance are those caused by suprarenal aortic coarctation or constriction of one of the renal arteries and removal of the contralateral kidney (eg, one-kidney, one-clip Goldblatt hypertension). Immediately after constriction of the renal artery or aortic coarctation, renal blood flow is reduced, and there is a rapid increase in renin secretion and transient sodium retention. Within a few days, sodium excretion returns to normal, and sodium balance is reestablished. If sodium and fluid intakes are normal, renin secretion also returns to normal in the established phase of hypertension. At this point, most indices of renal function are nearly normal, including blood pressure distal to the stenosis if the constriction is not too severe. The rate at which renal function returns to normal and time course for development of hypertension depend the severity of the stenosis, the sodium intake, and the rate of Ang II formation.18
How do these experimental models of increased preglomerular resistance relate to human hypertension, other than the obvious conditions of aortic coarctation or renal artery stenosis? When Goldblatt first produced hypertension in dogs by constricting the main renal artery, his goal was to develop an experimental model that mimicked the pathologic changes found in human essential hypertension.20 Goldblatt noted that many hypertensive patients at autopsy had nephrosclerosis and hypothesized that essential hypertension might be caused by diffuse renal arteriolar sclerosis, particularly in preglomerular vessels. Presumably, functional or pathologic increases in preglomerular resistance at other sites besides the main renal arteries, such as the interlobular arteries or afferent arterioles, could increase blood pressure through the same mechanisms as activated by clipping the renal artery. For example, widespread structural increases in afferent arteriolar resistance (eg, nephrosclerosis) or functional increases in resistance caused by excessive activation of the SNS or high levels of catecholamines (eg, pheochromocytoma) would also cause hypertension through the same mechanisms as constriction of the main renal artery.
Some have questioned whether Goldblatt hypertension is relevant to human essential hypertension because of the observation that many hypertensive patients do not have obvious renal ischemia and have nearly normal renal blood flow and GFR. However, as previously discussed, there is little indication of renal ischemia even in experimental Goldblatt hypertension after compensatory increases in blood pressure have occurred.
It is interesting to note that some patients with primary hypertension have the same characteristics seen in the one-kidney, one-clip Goldblatt model of hypertension, including nearly normal GFR and plasma renin activity, a parallel shift of pressure natriuresis to higher blood pressure, and a relatively salt-insensitive form of hypertension.18,21 Indeed, studies in hypertensive patients show that drug therapy that decreases preglomerular resistance, such as calcium channel blockers, causes a parallel shift of pressure natriuresis toward lower blood pressure.22 Thus, primary hypertension in some patients may be caused by functional or pathologic increases in preglomerular resistance. This is almost certainly the case in patients who have severe atherosclerotic lesions in the renal blood vessels.
In the two-kidney, one-clip Goldblatt model of hypertension and in patients with a stenosis in only one renal artery, there is a nonhomogeneous increase in preglomerular resistance with ischemia occurring in nephrons of the clipped or stenotic kidney; nephrons in the contralateral nonclipped or nonstenotic kidney have normal or increased single-nephron blood flow and GFR. Whereas the underperfused clipped kidney secretes large amounts of renin, the untouched kidney secretes very little renin.18,19
An important distinction between a generalized increase in preglomerular resistance and patchy, nonhomogeneous increases in preglomerular resistance is the evolution of renal injury associated with hypertension. When there is a generalized, homogeneous increase in preglomerular resistance, the glomeruli are protected from the damaging effects of increased blood pressure. In the two-kidney, one-clip model, however, the glomeruli of the untouched kidney are subjected to the full effects of increased blood pressure. With prolonged hypertension, pathologic changes in the untouched kidney add to the impairment of overall renal excretory capability. At this stage, removal of the clipped kidney only partially restores arterial pressure to normal. However, removal of the contralateral untouched kidney and unclipping the stenotic kidney normalizes blood pressure. Thus, chronic exposure to high blood pressure in the untouched kidney causes structural changes as well as functional changes that contribute to the progression of hypertension in this model.
The relevance of experimental models of nonhomogeneous increases in preglomerular resistance to human hypertension is obvious when there is stenosis of only one renal artery with the contralateral kidney being initially unaffected. If the contralateral nonstenotic kidney eventually becomes injured as a result of being exposed to high blood pressure, this kidney may also contribute to the hypertension.
Some patients with essential hypertension may have patchy nephrosclerosis within each kidney also providing another clinical counterpart to the two-kidney, one-clip Goldblatt model of hypertension. In these instances, the ischemic nephrons secrete large amounts of renin, and the nonischemic nephrons may initially have increased single-nephron GFR. The combined effects of hypertension and hyperfiltration, however, may eventually damage the nephrons that were not initially ischemic, leading to progressive nephron loss.
Reducing the glomerular capillary filtration coefficient (Kf) initially lowers GFR and sodium excretion while stimulating renin release and causing vasodilation of afferent arterioles via a macula densa feedback.18,19 The sodium retention and increased Ang II formation increase arterial pressure, which then helps to restore GFR and renin release to normal. After these compensations, the main persistent abnormalities of kidney function are reduced filtration fraction, increased glomerular hydrostatic pressure, and increased renal blood flow.
Unfortunately, compensatory increases in blood pressure and glomerular hydrostatic pressure, which offset a decrease in Kf and restore sodium excretion to normal, may also lead to additional renal dysfunction over a period of years by causing further glomerular injury; the gradual injury and loss of glomeruli further reduce Kf and require additional increases in blood pressure to maintain normal water and electrolyte balances. Such a sequence may initiate progressive kidney damage and worsening of hypertension.
The clinical counterparts of this sequence may be found in hypertension caused by glomerulonephritis or by other conditions that cause thickening and damage to the glomerular capillary membranes, such as chronic diabetes mellitus.23
A factor that contributes to salt sensitivity of blood pressure in some hypertensive patients is loss of functional nephrons. Complete loss of nephrons (eg, surgical reduction of kidney mass or unilateral nephrectomy) in the absence of other abnormalities usually does not lead to significant hypertension.18,19 In contrast, loss of functional nephrons because of ischemia or infarction of renal tissue usually causes marked hypertension that is initially caused by increased renin secretion and then is eventually mediated by additional abnormalities, such as immunologic and renal injury, in the established phase of the hypertension.24
One might suspect that reductions in nephron number should impair renal excretory capability and cause hypertension regardless of whether the loss was associated with renal ischemia. Yet experimental studies show that surgical removal of large amounts of the kidney, to the point that uremia occurs, rarely causes severe hypertension as long as sodium intake is normal.18,25 The reason hypertension does not develop is that glomerular filtration and tubular reabsorption capability are proportionally reduced so that balance between filtration and reabsorption can be maintained without major adaptive changes in blood pressure.
Reducing the number of functional nephrons, however, does make the kidneys susceptible to additional insults that impair their function or to additional challenges of sodium homeostasis. Thus, hypertension associated with excess mineralocorticoids is much more severe after reducing kidney mass. Likewise, the kidney’s ability to increase sodium excretion in response to the additional challenge of high sodium intake is accompanied by much larger increases in blood pressure when kidney mass is reduced.18,19
With the loss of entire nephrons, the surviving nephrons must excrete greater amounts of sodium and water to maintain balance. This is achieved by increasing GFR and decreasing reabsorption in the remaining nephrons, resulting in increased sodium chloride delivery to the macula densa and suppression of renin release. This, in turn, impairs the kidney’s ability to further decrease renin secretion during high sodium intake, and blood pressure becomes very salt sensitive.
Nephron loss may also initiate compensatory changes that damage the surviving nephrons.23 For example, over long periods of time, renal vasodilation and increased single-nephron GFR may lead to glomerulosclerosis and reductions in Kf. These pathologic changes, in addition to the loss of functional nephrons, may eventually impair pressure natriuresis sufficiently to cause severe hypertension.
What is the relevance of experimental models produced by surgically removing kidney mass to human hypertension? With normal aging, especially after age 40 to 50 years, there is gradual nephron loss that is accelerated by renal diseases, such as glomerulonephritis, diabetes mellitus, or long-standing hypertension. Thus, even though hypertension may not begin with nephron loss, chronic elevations in glomerular pressure and other metabolic abnormalities that are often associated with hypertension may eventually cause progressive nephron loss that amplifies the hypertension and makes blood pressure more salt sensitive.
The experimental model of surgical reduction of kidney mass discussed above should not be confused with the model of partial renal infarction hypertension produced by tying off branches of the renal artery, the so-called 5/6 ablation model. This model is usually produced by removing one kidney and obstructing two of the three branches of the renal artery of the remaining kidney. In the infarction model, hypertension develops even without a high sodium intake because of ischemia of the surviving nephrons, activation of the RAS, and immune-mediated injury of the kidney.24 The 5/6 renal ablation hypertension is a model of severe patchy renal ischemia with characteristics similar to that described for the two-kidney, one-clip Goldblatt model or nonhomogeneous patchy glomerulosclerosis. The clinical counterpart of this model occurs with partial renal infarction caused by septic emboli, thrombus, trauma, or sometimes after corrective surgery for renal artery stenosis.
Hypertension can also be caused by factors that increase renal tubular sodium reabsorption, such as excessive levels of mineralocorticoids or Ang II. The severity of hypertension depends on the degree to which tubular reabsorption is stimulated and on other factors, such as the functional kidney mass and sodium intake.18,19 With nephron loss or high sodium intake, the hypertensive potency of mineralocorticoids or Ang II is greatly enhanced.
One feature of hypertension caused by increased distal or collecting tubular reabsorption is that it is usually salt sensitive, with increased sodium intake exacerbating the hypertension. Increased reabsorption at sites beyond the macula densa, such as the distal tubules and collecting tubules, elicits chronic increased sodium chloride delivery to the macula densa, which, in turn, suppress renin secretion.14,18,19 Reduction of renin secretion to very low levels, characteristic of disorders associated with increased distal or collecting tubular reabsorption, prevents further suppression of Ang II formation during high sodium intake, making blood pressure salt sensitive.
Another feature of hypertension caused by increased tubular reabsorption (as occurs with excess aldosterone secretion) is that it is often associated with extracellular volume expansion. However, the initial volume expansion and increased cardiac output usually subside because of pressure natriuresis and total peripheral resistance increases because of the tissue blood flow autoregulatory mechanisms discussed previously. When increased tubular reabsorption is also associated with marked peripheral vasoconstriction, such as occurs with very high levels of Ang II, the degree of volume expansion depends on the relative effects of the vasoconstrictor on the peripheral blood vessels and the renal blood vessels.18,19 With severe peripheral vasoconstriction and decreased vascular capacitance, relatively small amounts of volume retention can lead to marked substantial hypertension.
An increase in tubular reabsorption occurring before the macula densa (eg, in the proximal tubules or loop of Henle) usually results in a salt-insensitive form of hypertension. Increased tubular reabsorption before the macula densa tends to increase renin secretion and elicits a compensatory renal vasodilation that raises GFR and renal plasma flow. However, as hypertension develops, macula densa sodium chloride delivery and renin secretion return to nearly normal, and the RAS is fully capable of responding to additional challenges such as increased sodium intake. Therefore, high sodium intake is accompanied by appropriate suppression of Ang II formation, which permits sodium balance to be maintained with only small increases in blood pressure.14,18 Nevertheless, the pressure natriuresis mechanism is shifted to higher blood pressure, parallel to the normal curve, and the severity of the hypertension depends on the degree to which tubular reabsorption is increased.
Significance of Salt-Sensitive Hypertension
Salt sensitivity of blood pressure is a quantitative phenotype, rather than following a bimodal categorization of salt sensitive or salt insensitive, and there is considerable heterogeneity of blood pressure responses to changes in sodium intake in normotensive as well as in hypertensive individuals.26
Although various methods have been used to assess salt sensitivity, none are widely used in clinical practice. Most salt-sensitivity protocols involve relatively short-term changes in sodium intake, usually over a few days. Weinberger et al26 defined salt sensitivity as a 10 mm Hg or greater change in mean blood pressure from the level measured after a 4-hour infusion of 2 L of normal saline compared with the level measured the morning after 1 day of a low-sodium (10 mmol) diet and administration of three doses of furosemide. With this definition, 51% of hypertensive and 26% of normotensive subjects were found to be salt sensitive.26 However, there has been little effort to determine the repeatability of salt sensitivity in the same persons over long periods of time (years), and it is not known whether short-term blood pressure responses reliably predict the long-term effects of changes in salt intake.
From clinical observations, it is clear that many demographic and pathophysiologic conditions are associated with salt sensitivity. Older individuals are usually more salt sensitive than young people, and African Americans are often more salt sensitive than whites. However, there are many exceptions to these generalizations, and considerable heterogeneity exists in the blood pressure responses to changes in salt intake.
Genetic factors independent of ethnicity have also been linked to salt sensitivity of blood pressure, especially monogenic disorders that increase distal and collecting tubule sodium reabsorption or that cause excess secretion of sodium-retaining hormones (eg, mineralocorticoids).27 Also, diabetes mellitus, renal diseases that cause nephron loss, and abnormalities of the RAS are all associated with increased salt sensitivity of blood pressure.19,28 Most of these examples appear to share two common pathways to salt sensitivity of blood pressure: loss of functional nephrons or reduced responsiveness of the RAS.
Figure 69–7 shows the effect of surgically reducing kidney mass on salt sensitivity in dogs. As long as sodium intake was normal, surgical reduction of kidney mass by 25%, or even as much as 70%, did not markedly alter blood pressure.25 However, after loss of kidney mass, blood pressure became exquisitely sensitive to changes in sodium intake, and with high sodium intake, blood pressure increased by approximately 40 mm Hg.
Figure 69–7

The effect of reducing kidney mass on salt sensitivity of blood pressure in dogs. Note that as long as sodium chloride intake was normal, surgical reduction of kidney mass by 25% or even 70% did not markedly alter arterial pressure. However, after loss of kidney mass, blood pressure became exquisitely sensitive to high sodium chloride intake. Redrawn with permission from Langston et al.25
How is this experimental study conducted in dogs relevant to human hypertension? Even in normal, healthy people, there is a gradual reduction in nephron number of about 10% per decade after age 40 years.29 At age 70 years, the average healthy individual typically has at least a 30% decrease in the number of nephrons compared with a young adult, a factor that may contribute to the increase in blood pressure with age in industrialized societies. When a person has an underlying renal disease, hypertension, or diabetes, the loss of nephrons with aging is greatly exacerbated. Other less common causes of nephron loss include primary renal diseases such as glomerulonephritis, acquired renal disease caused by analgesic abuse, uncontrolled diabetes mellitus, and developmental causes because of poor maternal nutrition or placental ischemia.
Figure 69–8 shows the importance of changes in Ang II formation in maintaining blood pressure relatively constant during variations in salt intake from a very low level of 5 mmol/d up to 80, 240, and 500 mmol/d for 8 days at each level. In normal dogs with a functional RAS, there were only small increases in blood pressure associated with this 100-fold range of sodium intakes.30,31 However, when Ang II was infused at a low level that initially had little effect on blood pressure but that prevented Ang II from being suppressed as sodium intake was raised, blood pressure became very salt sensitive. After blockade of Ang II formation, blood pressure also became salt sensitive, although pressure was maintained at a much lower level, especially when sodium intake was low.31 Thus, one of the major functions of the RAS is to permit wide variations in intake and excretion of sodium without large fluctuations in blood pressure that would otherwise be needed to maintain sodium balance.
Figure 69–8
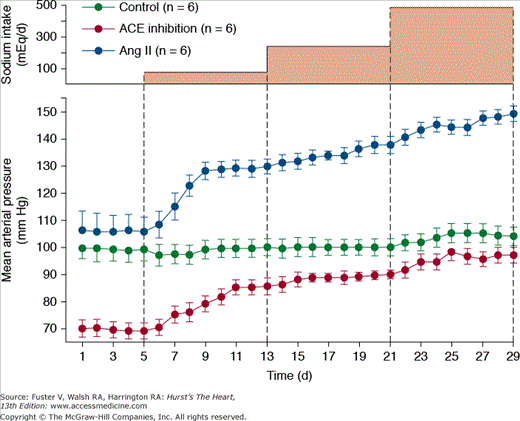
Changes in mean arterial pressure during chronic changes in sodium intake in normal control dogs, after angiotensin-converting enzyme (ACE) inhibition, or after angiotensin Ang II infusion (5 ng/kg/min) to prevent Ang II from being suppressed when sodium intake was raised. Redrawn with permission from Hall et al.31
What clinical conditions can lead to reduced responsiveness of the RAS? As discussed previously, focal nephrosclerosis or patchy preglomerular vasoconstriction, as occurs with renal infarction, leads to increased renin secretion in ischemic nephrons and very low levels of renin release by overperfused nephrons.18,19,32 Thus, in ischemic, as well as overperfused, nephrons, the ability to adequately suppress renin secretion during high salt intake is impaired.
Another cause of reduced responsiveness of the RAS is increased distal and collecting tubular sodium reabsorption as occurs with mineralocorticoid excess or mutations that increase distal and collecting tubule reabsorption (eg, Liddle syndrome). In these conditions, excess sodium retention causes almost complete suppression of renin secretion, resulting in an inability to further decrease renin release during high sodium intake. Consequently, blood pressure becomes very salt sensitive.
What is the clinical significance of salt sensitive blood pressure besides the obvious fact that it provides insight into the pathogenesis of hypertension and it indicates which patients may benefit most from reduction of salt intake? Some studies suggest that salt sensitivity predicts which patients are at greatest risk for hypertensive target-organ injury. Salt-sensitive forms of hypertension are often associated with glomerular hyperfiltration and increased glomerular hydrostatic pressure that is further amplified by the hypertension (see Table 69–2)19; together, the hypertension and renal hyperfiltration promote glomerular injury and may eventually cause loss of nephron function. Clinical studies support this concept and demonstrate that whereas salt-sensitive individuals typically have increased glomerular hydrostatic pressure and albumin excretion when given a salt load, salt-resistant individuals have lower glomerular hydrostatic pressure and less urinary albumin excretion.33
There is also evidence that salt-sensitive subjects may die earlier than individuals who are salt resistant. In a study by Weinberger et al34 in which individuals were followed for more than 20 years, normotensive individuals with increased salt sensitivity died almost at the same rate as hypertensive individuals and much faster than salt-resistant individuals who were normotensive. Whether this increased mortality was related to blood pressure effects of salt or to other effects is still unclear. It is also not known whether long-term high salt intake, lasting over many years, may cause a person who is initially “salt insensitive” to become “salt sensitive” as a consequence of gradual renal injury.
Neurohumoral Mechanisms of Hypertension
Although impaired renal pressure natriuresis plays a central role in all forms of hypertension studied thus far, not all disorders of pressure natriuresis originate within the kidneys. Inappropriate activation of the multiple antinatriuretic hormone systems (eg, Ang II, aldosterone) that normally regulate sodium excretion or deficiency of natriuretic influences (eg, atrial natriuretic peptide [ANP],NO) on the kidneys can impair pressure natriuresis and cause chronic hypertension. Likewise, excessive activation of the SNS plays a major role in elevating blood pressure in many hypertensive patients. The following sections discuss the multiple neural, hormonal, and autacoid mechanisms that contribute to long-term blood pressure regulation, their actions on the kidneys, and their potential roles in hypertension.
The SNS is a major short- and long-term controller of blood pressure. Sympathetic vasoconstrictor fibers are distributed to almost all of regions of the vasculature, as well as to the heart, and activation of the SNS can raise blood pressure within a few seconds by causing vasoconstriction, increased cardiac pumping capability, and increased heart rate. Conversely, sudden inhibition of SNS activity can decrease blood pressure to as low as half normal within less than 1 minute. Therefore, changes in SNS activity, caused by various reflex mechanisms, CNS ischemia, or by activation of higher centers in the brain, provide powerful and rapid, moment-to-moment regulation of blood pressure.
The SNS also plays an important role in long-term regulation of blood pressure and in the pathogenesis of hypertension, in large part by activation of the renal sympathetic nerves.35 There is extensive innervation of the renal blood vessels, the juxtaglomerular apparatus, and the renal tubules and excessive activation of these nerves causes sodium retention, increased renin secretion, and impaired renal pressure natriuresis.35 Except for extreme circumstances, such as severe hemorrhage or other conditions associated with marked circulatory depression, activation of the renal sympathetic nerves is usually not great enough to cause marked reductions in renal blood flow or GFR. However, even mild increases of the renal sympathetic activity stimulate renin secretion and sodium reabsorption in multiple segments of the nephron, including the proximal tubule, the loop of Henle, and more distal segments.35 Thus, the renal nerves provide a mechanism by which the various reflex mechanisms and higher CNS centers may contribute to long-term regulation of blood pressure.
The preganglionic neurons that synapse with the renal sympathetic postganglionic fibers are located in the lower thoracic and upper lumbar segments of the spinal cord and receive multiple inputs from various regions of the brain, including the brainstem, forebrain, and cerebral cortex. These complex neural circuits provide multiple pathways by which neural reflexes and higher CNS centers can influence renal SNS activity and chronic regulation of blood pressure.36,37
Evidence for a role of the renal nerves in hypertension comes from multiple studies showing that renal denervation reduces blood pressure in some models of experimental hypertension.35 For example, complete renal denervation attenuates the development of hypertension in spontaneously hypertensive rats35 as well as in obese hypertensive dogs.38 Renal denervation may also delay or attenuate increased blood pressure in other forms of experimental hypertension, although some studies have not found an important role for the renal nerves in various forms of secondary hypertension. In Ang II hypertension, for example, decreased renal sympathetic activity appears to attenuate the rise in blood pressure.39
Human primary hypertension, especially when associated with obesity, is often associated with increased renal sympathetic activity.40 Bilateral renal denervation in humans using a percutaneous, catheter-based radiofrequency method to selectively ablate the nerves that run along the renal arteries caused highly significant reductions in blood pressure in patients who were resistant to the usual antihypertensive drugs.41,42 No attenuation of blood pressure reduction was detected over several months of follow-up, suggesting the absence of nerve fiber regrowth. However, longer periods of follow-up will be needed to determine if the renal nerves eventually regrow and reinitiate increases in blood pressure as has been observed in experimental animal models of renal denervation.43
Although the mechanisms that cause activation of renal sympathetic nerves in primary hypertension or in most experimental models are still unclear, we will briefly discuss three that have attracted the interest of many researchers.
The importance of the arterial baroreceptors in buffering moment-to-moment changes in blood pressure is clearly evident in baroreceptor-denervated animals in which there is extreme variability of blood pressure associated with normal daily activities.44 After baroreceptor denervation, blood pressure increases to very high levels or decreases to low levels with normal daily activities, although the average 24-hour mean arterial pressure is not markedly altered.
The arterial baroreceptors clearly provide a powerful means for moment-to-moment regulation of arterial pressure, but their role in long-term blood pressure regulation is controversial. Some studies suggest that the baroreceptors are relatively unimportant in chronic regulation of blood pressure because they tend to reset within a few days to the level of blood pressure to which they are exposed.44 In most forms of chronic hypertension, the arterial baroreflexes are reset to higher blood pressures. To the extent that resetting of baroreceptors occurs, this would attenuate their potency as a long-term controller of blood pressure.
Other experimental studies, however, suggest that the baroreceptors do not completely reset and therefore may contribute to long-term blood pressure regulation. With prolonged increases in arterial pressure, the baroreflexes may contribute to reductions in renal sympathetic activity and promote sodium and water excretion.39 This in turn may attenuate the increase in arterial blood pressure. Thus, impairment of baroreflexes may cause increased lability of blood pressure in hypertension and may fail to attenuate the increase in blood pressure caused by other disturbances. However, there is currently little evidence that primary disturbances of baroreceptor function play a major role in causing chronic hypertension.
Nevertheless, experimental studies in dogs indicate that chronic electrical stimulation of carotid sinus baroreceptors reduces blood pressure in experimental models of hypertension.45,46 In humans with hypertension that was resistant to drug treatment, electrical stimulation of baroreceptors also caused significant reductions in blood pressure.47 These observations are consistent with the hypothesis that strong activation of baroreceptors can have long-term influences on blood pressure. However, the primary role of arterial baroreceptors in hypertension, as in normotension, is to buffer deviations in blood pressure from the setpoint determined by renal pressure natriuresis.
Increased blood pressure lability associated with baroreflex dysfunction, however, is accompanied by periodic large increases in blood pressure that may cause gradual renal injury and eventually lead to chronic hypertension. Studies in experimental animals show, for example, that baroreceptor-denervated animals have significant structural changes in the kidneys, including glomerular injury.48
Acute physiologic stresses, including pain, exercise, exposure to cold, and mental stress, can all lead to increased SNS activity and transient hypertension. It is also widely believed, however, that chronic stress may lead to long-term increases in blood pressure. Support for this concept comes largely from a few epidemiologic studies showing that air traffic controllers, lower socioeconomic groups, and other groups who are believed to lead more stressful lives also have increased prevalence of hypertension.49 There is limited evidence, however, for a direct cause-and-effect relationship between psychosocial stress and chronic hypertension. Nevertheless, there is widespread belief by many researchers and by the general public that stress is an important cause of hypertension in humans.
As discussed in more detail later, excess weight gain appears to be a major cause of human primary hypertension. The mechanisms responsible for obesity hypertension appear to be closely linked to increased renal SNS activity.14,50,51 Obese persons have elevated SNS activity in various tissues, including the kidneys and skeletal muscle, as assessed by microneurography, tissue catecholamine spillover, and other methods.14,40 Studies in experimental animals and humans indicate that combined α- and β-adrenergic blockade markedly attenuates the hypertension associated with obesity.16,50,52 Moreover, the renal sympathetic efferent nerves mediate much of the chronic effects of SNS activation on blood pressure in obesity as bilateral renal denervation greatly attenuates the sodium retention and hypertension in obese dogs.38 Thus, obesity increases renal sodium reabsorption, impairs pressure natriuresis, and causes hypertension partly by increasing renal SNS activity.
The RAS is perhaps the most powerful hormone system for regulating body fluid volumes and blood pressure as evidenced by the effectiveness of various RAS blockers in reducing blood pressure in normotensive and hypertensive subjects. Although the RAS has many components, its most important effects on blood pressure regulation are exerted by Ang II, which participates in both short- and long-term control of arterial pressure.
Ang II is a powerful vasoconstrictor and helps maintain blood pressure in conditions associated with acute volume depletion (eg, hemorrhage), sodium depletion, or circulatory depression (eg, heart failure). The long-term effects of Ang II on blood pressure, however, are closely intertwined with volume homeostasis through direct and indirect effects on the kidneys.
When the RAS is fully functional, the chronic renal pressure natriuresis curve is steep, and sodium balance can be maintained over a wide range of intakes with minimal changes in blood pressure (Fig. 69–9). One reason for the effectiveness of the normal pressure natriuresis mechanism is that Ang II levels are suppressed during high sodium intake and increased when sodium intake is restricted, thereby adjusting renal sodium excretion appropriately without the need for large changes in blood pressure to maintain sodium balance.
Figure 69–9
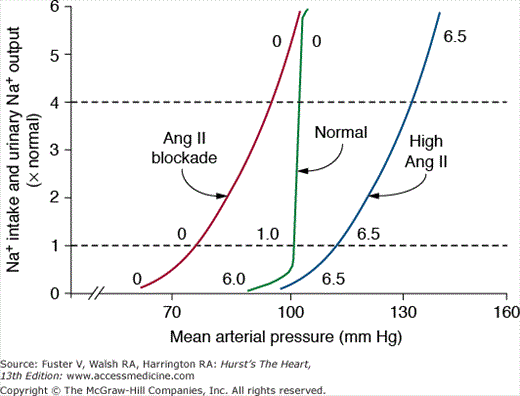
Steady-state relationships between arterial pressure and sodium intake and excretion under normal conditions with a fully functional renin–angiotensin system, after blockade of angiotensin II (Ang II) formation with an angiotensin-converting enzyme inhibitor, and after Ang II infusion at a low dose to prevent Ang II levels from being suppressed when sodium intake was increased. The numbers along the curves are estimated Ang II levels expressed as times normal. Redrawn with permission from Hall et al.31
Blockade of the RAS, with Ang II receptor blockers (ARBs) or angiotensin- converting enzyme (ACE) inhibitors, increases renal excretory capability so that sodium balance can be maintained at reduced blood pressure.19 However, blockade of the RAS also reduces the slope of pressure natriuresis and makes blood pressure salt sensitive.30 Thus, the effectiveness of RAS blockers in lowering blood pressure is greatly diminished by high salt intake; conversely, reducing sodium intake or addition of a diuretic improves the effectiveness of RAS blockers in reducing blood pressure.
Inappropriately high levels of Ang II reduce renal excretory capability and impair pressure natriuresis, thereby reducing the slope and necessitating increased blood pressure to maintain sodium balance. The mechanisms that mediate the potent antinatriuretic effects of Ang II include direct and indirect effects to increase tubular reabsorption as well as renal hemodynamic effects.30
Physiologic activation of the RAS usually occurs as a compensation for conditions that cause volume depletion or underperfusion of the kidneys, such as sodium depletion, hemorrhage, or heart failure. Increased formation of Ang II helps to restore renal perfusion by causing salt and water retention, which in turn tends to prevent reductions in blood pressure. Ang II causes salt and water retention by increasing renal sodium reabsorption through stimulation of aldosterone secretion, by direct effects on epithelial transport, and by hemodynamic effects.
Ang II–mediated constriction of efferent arterioles reduces renal blood flow and peritubular capillary hydrostatic pressure and increases peritubular colloid osmotic pressure as a result of increased filtration fraction.30 These changes, in turn, increase the driving force for fluid reabsorption across tubular epithelial cells. Reductions in renal medullary blood flow caused by efferent arteriolar constriction or by direct effects of Ang II on the vasa recta may also enhance reabsorption in the loop of Henle and collecting ducts.30
Ang II also directly stimulates tubular sodium reabsorption. This effect occurs at very low Ang II concentrations and is mediated in by actions on the luminal and basolateral membranes.30,53 In the proximal tubules, Ang II stimulates the Na+-H+ exchanger on the luminal membrane and increases sodium-potassium ATPase activity, as well as sodium bicarbonate cotransport on the basolateral membrane (Fig. 69–10).53,54 These effects are partly mediated by inhibition of adenyl cyclase and increased phospholipase C activity.
Figure 69–10
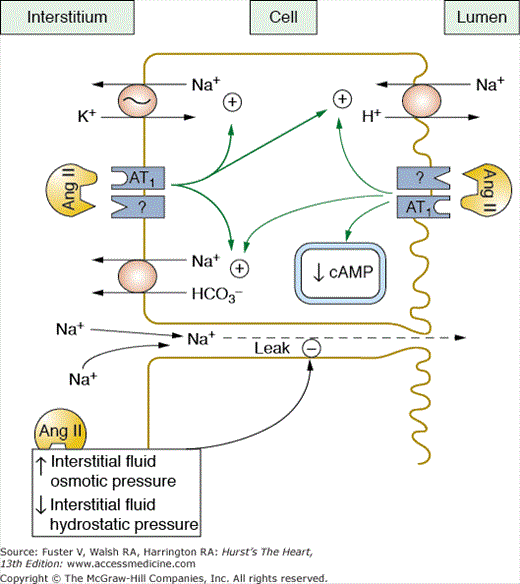
Angiotensin Ang II increases proximal tubular reabsorption by binding to receptors on the luminal and basolateral membranes and stimulating Na+/H+ antiporter, Na+/HCO3 cotransport, and Na+/K+ adenosine triphosphatase (ATPase) activity. Ang II also increases reabsorption by increasing interstitial fluid colloid osmotic pressure and decreasing interstitial fluid hydrostatic pressure. Redrawn with permission from Hall et al.53
Sodium reabsorption in the loop of Henle, macula densa, and distal nephron segments is also stimulated by Ang II. At physiologic concentrations, Ang II increases bicarbonate reabsorption in the loop of Henle and stimulates Na+-K+-2 Cl transport in the medullary thick ascending loop of Henle.53,54 Ang II stimulates multiple ion transporters in the distal parts of the nephron, including H+-ATPase activity, as well as epithelial sodium channel activity in the cortical collecting ducts.53,54
Ang II is a powerful renal vasoconstrictor but in most physiologic conditions, the constriction is confined mainly to the postglomerular efferent arterioles. For example, efferent arteriolar constriction by Ang II acts in concert with other autoregulatory mechanisms, such as tubuloglomerular feedback (TGF) and myogenic activity, to prevent excessive reductions in GFR when kidney perfusion is threatened.19,30 In these cases, administration of ARBs or ACE inhibitors may actually reduce GFR further, even though renal blood flow is preserved. The impairment of GFR after RAS blockade is caused, in part, by inhibition of the constrictor effects of Ang II on efferent arterioles as well as reduced blood pressure.30
The constrictor effects of Ang II on efferent arterioles are especially important in patients with renal artery stenosis or sodium depletion or with heart failure who may have substantial decreases in GFR when treated with RAS blockers.30
The weak constrictor action of Ang II on preglomerular vessels is related, in part, to selective protection of these vessels by autacoid mechanisms such as prostaglandins (PGs) or endothelial-derived NO.30 When the ability of the kidneys to produce these autacoids is impaired by treatment with nonsteroidal anti-inflammatory drugs (NSAIDs) or by chronic vascular disease (eg, atherosclerosis), increased levels of Ang II may reduce GFR by constricting afferent arterioles.
Although blockade of the vasoconstrictor effects of Ang II on the efferent arterioles may cause a further decline in GFR in ischemic nephrons, RAS blockade may be beneficial when nephrons are hyperfiltering, especially if Ang II is not appropriately suppressed. For example, in diabetes mellitus and certain forms of hypertension associated with glomerulosclerosis and nephron loss, Ang II blockade, by decreasing efferent arteriolar resistance and arterial pressure, lowers glomerular hydrostatic pressure and attenuates glomerular hyperfiltration.53,54 Clinical and experimental studies indicate that RAS blockers are more effective than other antihypertensive agents in preventing glomerular injury, even with similar reductions in blood pressure55-57; this appears to be partly caused by a greater reduction in glomerular hydrostatic pressure as a result of vasodilation of efferent arterioles after blockade of the RAS.
Ang II has been suggested to cause injury to the kidneys and other organs through direct actions in addition to its hemodynamic effects. Much of the evidence supporting this hypothesis comes from in vitro studies, often using supraphysiologic concentrations of Ang II. Although in vivo studies have demonstrated greater renal protective effects of RAS blockers compared with other antihypertensive drugs, decreases in glomerular hydrostatic pressure because of efferent arteriolar vasodilation may have contributed to these beneficial effects. In studies in which blood pressure was measured very accurately, using 24-hour telemetry, the renal protective effects of RAS blockade appear to be largely a result of reductions in blood pressure.58
An observation that is difficult to reconcile with the concept that Ang II directly mediates target organ injury, independent of blood pressure, is the finding that physiologic activation of the RAS is not associated with vascular or renal injury as long as the blood pressure is not elevated. For example, sodium depletion does not cause renal, cardiac, or vascular injury despite marked increases in renal Ang II levels. Also, the clipped kidney of the two-kidney, one-clip Goldblatt model of hypertension is exposed to very high Ang II levels but is protected from increased arterial pressure by the clip on the renal artery and has no visible injury as long as the stenosis is not too severe. However, the nonclipped kidney, exposed to lower Ang II concentrations but higher blood pressure, has marked focal segmental glomerular sclerosis as well as tubulointerstitial changes characteristic of hypertension.59
Convincing evidence in this controversial area of research comes from the observations of Crowley and colleagues, who studied the effects of chronic Ang II infusion in normal wild-type (WT) mice, in WT mice that received transplanted kidneys from AT1 receptor knock-out mice, and in AT1 receptor knock-out mice that received transplanted kidneys from normal WT mice.60 Chronic Ang II infusion in normal WT mice increased blood pressure and caused cardiac hypertrophy and fibrosis. However, in WT mice that received transplanted kidneys from AT1 receptor knock-out mice (ie, AT1 receptors were present in the heart and other organs but not in the kidneys), Ang II infusion did not chronically increase blood pressure or cause cardiac hypertrophy and fibrosis. In AT1 receptor knock-out mice that received transplanted kidneys from normal WT mice (ie, AT1 receptors were present only in the kidneys and not in the heart or other organs), Ang II infusion caused chronic hypertension as well as cardiac hypertrophy and fibrosis.60 These observations indicate that (1) the renal effects of Ang II, and not the peripheral vascular or other non-renal effects, mediate chronic increases in blood pressure and (2) in the absence of hypertension, Ang II does not cause cardiac hypertrophy or fibrosis. Thus, the hemodynamic effects appear to account for most of the target injury that occurs in Ang II–dependent hypertension.
Aldosterone is also a powerful sodium-retaining hormone and consequently has important effects on pressure natriuresis and blood pressure regulation. The primary sites of actions of aldosterone on sodium reabsorption are the principal cells of the distal tubules, cortical collecting tubules, and collecting ducts where aldosterone stimulates sodium reabsorption and potassium secretion. Aldosterone binds to intracellular mineralocorticoid receptors (MRs) and activates transcription by target genes which, in turn, stimulate synthesis or activation of the Na+-K+-ATPase pump on the basolateral epithelial membrane and activation of amiloride-sensitive sodium channels on the luminal side of the epithelial membrane.61 These effects are termed genomic because they are mediated by activation of gene transcription and require 60 to 90 minutes to occur after administration of aldosterone.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


