Chapter 7 Ion Channelopathies
Mechanisms and Genotype-Phenotype Correlations
Recent years have witnessed an explosion of knowledge contributing to the understanding of ion channelopathies associated with inherited cardiac arrhythmia syndromes that are responsible for the sudden death of infants, children, and young adults. These ion channelopathies are the consequences of genetic variations giving rise to primary electrical diseases, including long QT syndrome (LQTS), short QT syndrome (SQTS), and Brugada syndrome (BrS), as well as catecholaminergic ventricular tachycardia (VT) (Table 7-1).1–3 This review focuses on the molecular, genetic, cellular, and ionic mechanisms underlying the arrhythmogenesis associated with these syndromes and the genotype-phenotype correlation.
Brugada Syndrome
Arrhythmogenesis in BrS is believed to be the result of amplification of heterogeneities in the action potential characteristics among the different transmural cell types in the right ventricular (RV) myocardium.4,5 A decrease in sodium (Na+) or calcium (Ca2+) channel current, INa or ICa, or augmentation of any one of a number of outward currents, including rapidly activating delayed rectifier potassium (K+) current (IKr) or transient outward current (Ito), can cause preferential abbreviation of the right ventricular epicardial action potential; this, in turn, leads to the development of spatial dispersion of repolarization and, thus, the substrate and trigger for VT, which is usually polymorphic and less frequently monomorphic.6–12
BrS displays an autosomal dominant mode of inheritance. For many years, the only gene linked to BrS was SCN5A, the gene encoding for the α-subunit of the cardiac Na+ channel gene.6 However, recent evidence has shown that mutations in other genes are linked to the development of BrS.
BrS1, SCN5A
Mutations in SCN5A were the first to be associated with BrS.6 Over 293 mutations in SCN5A have now been linked to the syndrome.13 About three dozen of these have been studied in expression systems and shown to result in loss of function of the Na+ channel because of the following reasons: (1) failure of the sodium channel to express; (2) a shift in the voltage dependence and time dependence of INa activation, inactivation, or reactivation; (3) entry of the Na channel into an intermediate state of inactivation from which it recovers more slowly, or (4) accelerated inactivation of the Na+ channel.14–16 Premature inactivation of the Na+ channel has been observed at physiological temperatures but not at room temperature.17 Because this characteristic of the mutant channel is exaggerated at temperatures above the physiological range, it was suggested that the syndrome may be unmasked and that patients with BrS may be at an increased risk during a febrile state.17
BrS2, GPD1L
Weiss et al described a second locus on chromosome 3, close to but distinct from SCN5A, linked to the syndrome in a large pedigree in which the syndrome is associated with progressive conduction disease, a low sensitivity to procainamide, and a relatively good prognosis. The gene was recently identified as the glycerol-3-phosphate dehydrogenase 1-like (GPD1L) gene, and the mutation was found in this gene.18–20 Interestingly, it was also found that both GPD1L RNA and protein are abundant in the heart. Furthermore, the mutation was present in all affected individuals and absent in more than 500 control subjects. Coexpression studies of mutant GPD1L (A280V) with SCN5A in human embryonic kidney (HEK) cells resulted in a reduction in the magnitude of INa by approximately 50%.19 These studies provided evidence that mutations in GPD1L lead to a reduction in INa and cause BrS.19 Valdivia et al recently demonstrated that mutations in GPD1L related to BrS and sudden infant death syndrome (SIDS) cause a loss of enzymatic function, which results in glycerol-3-phosphate PKC-dependent phosphorylation of SCN5A at serine 1503 (S1503) through a GPD1L-dependent pathway. The direct phosphorylation of S1503 markedly decreases INa. These findings therefore show a function for GPD1L in cellular physiology and a mechanism linking mutations in GPD1L to sudden cardiac arrest. Because the enzymatic step catalyzed by GPD1L depends on nicotinamide adenine dinucleotide (NAD), this GPD1L pathway links the metabolic state of the cell to INa and excitability and may be important more generally in cardiac ischemia and heart failure.21
BrS3 and BrS4, CACNA1c and CACNB2b
The third and fourth genes associated with BrS were recently identified and were shown to encode the α1-subunit (CACNA1c) and the β-subunit (CACNB2b) of the L-type cardiac Ca channel.8 This new clinical entity, which exhibits electrocardiogram (ECG) and arrhythmic manifestations of both BrS and SQTS, was shown to be associated with loss of function mutations in the α1-subunit (CACNA1c) and the β-subunit (CACNB2b) of the L-type cardiac Ca2+ channel.8 Alterations in L-type Ca2+ current have been implicated in the development of BrS both clinically and experimentally.5,8 In both of those studies, the BrS phenotype was the result of a loss in peak ICa. More recently, a study identified a case of BrS in which the disease phenotype was observed as a result of accelerated inactivation of the L-type Ca2+ current without significantly affecting peak current.9 The accelerated inactivation was caused by a mutation in CACNB2b, which encodes the β-subunit of the cardiac L-type Ca2+ current. The carrier of this mutation exhibited ST-segment elevation in only one precordial lead and converted to a more typical BrS phenotype with a procainamide challenge. VT/VF (ventricular fibrillation) was inducible and subsequently detected on interrogation of the implanted implantable cardiac defibrillator (ICD), corroborating the diagnosis of a potentially life-threatening syndrome.
BrS5 and BrS7, SCN1B and SCN3B
Genes that encode cardiac channel β-subunit proteins have long been appealing candidates for the treatment of ion channelopathies such as BrS because of their significant role in modulating channel expression and function.22 The role of β1-subunits has been studied most extensively. Wild-type (WT) β1 coexpression has been reported to have no observable effect on SCN5A function, result in increased Na+ current density with no detectable effects on channel kinetics or voltage-dependence, modulate channel sensitivity to lidocaine blockade with subtle changes in channel kinetics and gating properties, and shift the voltage dependence of steady-state inactivation or alter the rate of recovery from inactivation.23–31 Coexpression of SCN5A with WT β3 results in either (1) increased current density, a depolarizing shift in the voltage-dependence of inactivation, and an increased rate of recovery from inactivation in Xenopus oocytes or (2) a hyperpolarizing shift of inactivation, slowed recovery from inactivation, and reduced late Na+ channel current.30,31
Mutations in SCN1B and SCN3B have recently been identified as the fifth and seventh genes associated with BrS. Mutations in β1-subunits (Navβ1 and Navβ1b) have been shown to be associated with combined BrS and cardiac conduction disease phenotype in humans.32 Another recent study by the authors of this chapter provided evidence that SCN3B is a BrS-susceptible gene.33 An L10P missense mutation in a highly conserved residue was shown to produce a major reduction in INa secondary to both functional and trafficking defects in cardiac Na+ channel expression. These results indicate that a mutation in the extracellular domain can impair trafficking of SCN5A to the membrane. These results suggest that WT β3 plays a role in facilitating SCN5A transport to the plasma membrane, since a mutation in the extracellular domain of β3 is capable of disrupting trafficking of SCN5A to the plasma membrane.33
BrS6, KCNE3
The role of the transient outward K+ current (Ito) is thought to be central to the development of BrS. This hypothesis comes from several lines of evidence. First, since BrS is characterized by ST-segment elevations in the right precordial leads, a more prominent Ito in RV epicardium has been suggested to underlie the much greater prevalence of the Brugada phenotype in males.34 The more prominent Ito causes the end of phase 1 of the RV epicardial action potential to repolarize to more negative potentials in tissue and arterially perfused wedge preparations obtained from male patients; this facilitates the loss of the action potential dome and the development of phase 2 re-entry and polymorphic VT. A link between mutations in genes responsible for the Ito current and the development of BrS was recently reported by Delpón and coworkers. KCNE3 was identified as the seventh gene associated with BrS.11 KCNE3 normally interacts with Kv4.3 to suppress Ito; and a mutation in KCNE3 was shown to result in a gain of function in Ito.11,35 Experimentally, the Brugada phenotype can be produced in arterially perfused wedge preparations by the Ito activator NS5806. This compound has been shown to increase peak Ito amplitude and slow inactivation in isolated cardiomyocytes, which results in a more prominent phase 1 repolarization and loss of the action potential (AP) dome in mid- and epicardial cells.36 The results of the study using the Ito activator are consistent with the clinical observations that an enhancement of Ito can lead to the development of BrS.
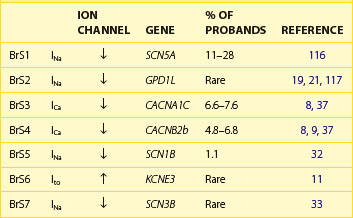
Table 7-2 lists the seven genotypes thus far associated for BrS and their yields. Four of the genes identified produced a loss of function of Na+ channel current; two led to a reduction in Ca2+ channel current; and one gene was associated with a gain of function of transient outward current. SCN5A mutations were identified in 11% to 28% of probands (average of 21%).13 Ca2+ channel mutations are found in approximately 12% to 15% of probands.8,37 Variations in the other genes were relatively rare.
Genotype-Phenotype Correlation
Patients with SCN5A-positive BrS exhibit conduction slowing characterized by prolonged PR intervals and QRS duration.38,39 PR interval and QRS duration are prolonged more prominently, and the QRS axis deviates more to the left with aging in those patients with BrS with SCN5A mutations. Smits et al observed significantly longer conduction intervals at baseline in patients with SCN5A mutations (PR and HV interval) and greater prolongation after the administration of Na+ channel blockers.38 These results concur with the observed loss of function of mutated BrS-related Na+ channels.
Patients with BrS who have calcium channel mutations are phenotypically distinct from those with mutations in other genes.8 A large fraction of CACNA1c– and CACNB2b-positive patients display a shorter-than-normal QTc interval (<360 ms) in addition to an ST-segment elevation in the right precordial leads, thus manifesting a combination of BrS and SQTS. Patients with mutations in Ca2+ channel genes also exhibit a diminished rate adaptation of QT interval.8,40
Mechanism of Arrhythmia in Brugada Syndrome
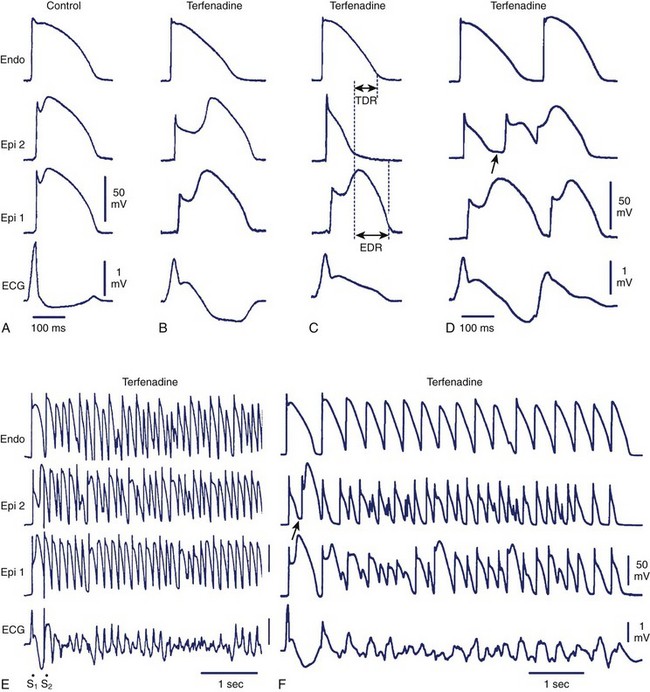
The arrhythmogenic substrate responsible for the development of extrasystoles and polymorphic VT in BrS is believed to be secondary to the amplification of heterogeneities intrinsic to the early phases (phase 1–mediated notch) of the action potential of cells residing in different layers of the right ventricular wall of the heart. Rebalancing of the currents active at the end of phase 1 is thought to underlie the accentuation of the action potential notch in the right ventricular epicardium, which is responsible for the augmented J wave and ST segment elevation associated with BrS (see41–43 for references). The presence of an Ito-mediated spike and dome morphology, or notch, in the ventricular epicardium but not in the endocardium creates a transmural voltage gradient that is responsible for the inscription of the electrocardiographic (ECG) J wave (Figure 7-1, A).5,44 The ST segment is normally isoelectric because of the absence of transmural voltage gradients at the level of the action potential plateau. Accentuation of the right ventricular action potential notch under pathophysiological conditions leads to exaggeration of transmural voltage gradients and thus to accentuation of the J wave or to J point elevation (Figure 7-1, B). If the epicardial action potential continues to repolarize before that of the endocardium, the T wave remains positive, giving rise to a saddleback configuration of the ST-segment elevation. Further accentuation of the notch is accompanied by a prolongation of the epicardial action potential causing it to repolarize after the endocardium, thus leading to inversion of the T wave. The down-sloping ST segment elevation, or accentuated J wave, observed in experimental wedge models often appears as an R, mimicking a right bundle branch block (RBBB) morphology of the ECG, largely because of early repolarization of the right ventricular (RV) epicardium, rather than major delays in impulse conduction in the right bundle.45 Despite the appearance of a typical Brugada sign, the electrophysiological changes shown in Figure 7-1, B, do not give rise to an arrhythmogenic substrate. The arrhythmogenic substrate may develop with a further shift in the balance of current leading to loss of the action potential dome at some epicardial sites but not others (Figure 7-1, C). A marked transmural dispersion of repolarization develops as a consequence, creating a vulnerable window, which can trigger a re-entrant arrhythmia when captured by a premature extrasystole. Because loss of the action potential dome in epicardium is generally heterogeneous, epicardial dispersion of repolarization develops as well. Conduction of the action potential dome from sites at which it is maintained to sites at which it is lost causes local re-excitation via phase 2 re-entry (Figure 7-1, D); this leads to the development of a closely coupled extrasystole that is capable of capturing the vulnerable window across the ventricular wall, thus triggering a circus movement re-entry in the form of VT/VF (Figures. 7-1, E and F).4,46 Support for these hypotheses comes from experiments involving the arterially perfused RV wedge preparations and from recent studies in which monophasic action potential (MAP) electrodes were positioned on the epicardial and endocardial surfaces of the right ventricular outflow tract (RVOT) in patients with BrS.4,5,41,47–51
Long QT Syndrome
LQTS is characterized by the appearance of long QT intervals on the ECG, an atypical polymorphic VT known as torsades de pointes (TdP), and a high risk for sudden cardiac death.52–54 A reduction of net repolarizing current secondary to loss of function of outward ion channel currents or a gain of function of inward currents underlies the prolongation of the myocardial action potential and QT interval that attend both congenital and acquired LQTS.55,56
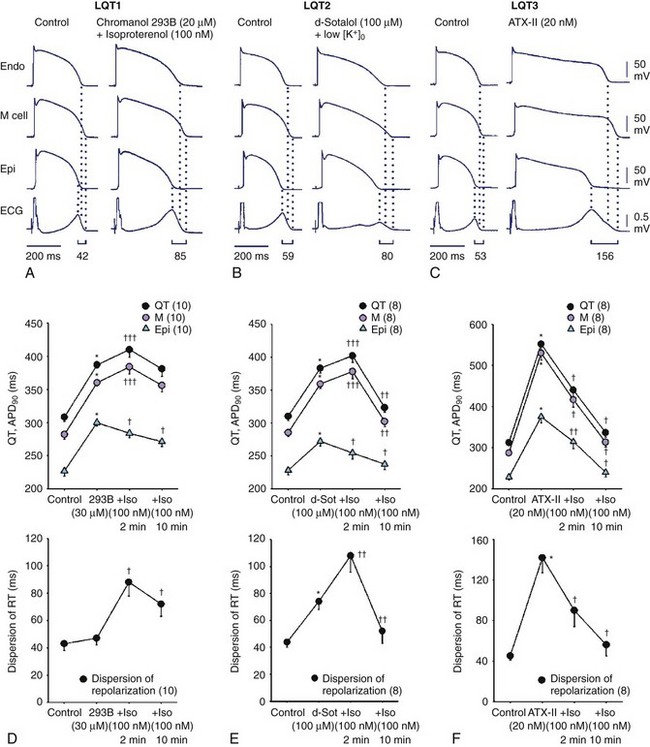
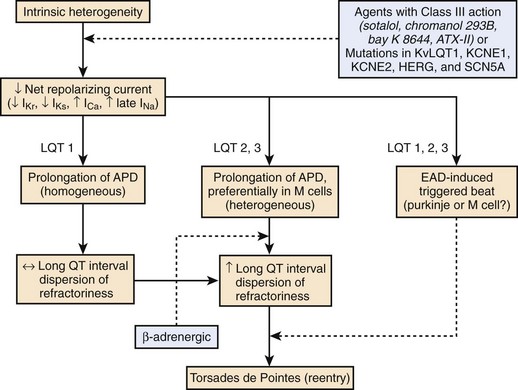
Here again, amplification of spatial dispersion of repolarization is thought to generate the principal arrhythmogenic substrate. The accentuation of spatial dispersion is secondary to an increase of transmural and trans-septal dispersion of repolarization. Early after-depolarization (EAD)–induced triggered activity also contributes to the development of the substrate and provides the triggering extrasystole that precipitates TdP arrhythmias observed under LQTS conditions (Figures 7-2 and 7-3).57,58 In vivo and in vitro models of LQTS have contributed to the understanding of the mechanisms involved in arrhythmogenesis.59,60 Models of the LQT1, LQT2, and LQT3 forms of LQTS have been developed by using arterially perfused left ventricular (LV) wedge preparations (see Figure 7-2) in canine models.61 These models have shown that in these three forms of LQTS, preferential prolongation of the M cell action potential duration (APD) leads to an increase in the QT interval as well as an increase in the transmural dispersion of repolarization (TDR), the latter providing the substrate for the development of spontaneous as well as stimulation-induced TdP.
LQT1, KCNQ1
LQT1 is the most prevalent of congenital LQTS.62 Loss of function of the slowly activating delayed rectifier (IKs) underlies congenital LQT1. Inhibition of IKs using chromanol 293B leads to uniform prolongation of APD in all three cell types (epicardial, endocardial, and M cell) in the wedge, causing little change in TDR. Although the QT interval is prolonged, TdP never occurs under these conditions, nor can it be induced. Addition of isoproterenol results in abbreviation of epicardial and endocardial APD, and the M cell APD either prolongs or remains the same. The dramatic increase in TDR provides the substrate for the development of spontaneous as well as stimulation-induced TdP.63 These results support the thesis that the problem with LQTS is not the long QT interval but, rather, the increase in TDR that often accompanies the prolongation of the QT interval. The combination of IKs block and β-adrenergic stimulation creates a broad-based T wave in the perfused wedge, similar to that observed in patients with LQT1. These findings provide an understanding of the great sensitivity of patients with LQT1 to sympathetic influences (see Figures 7-2, A and D).52,64
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


