Chapter 6 Genomics and Principles of Clinical Genetics
Elementary Understanding of Molecular Genetics
General Organization and Structure of the Human Genome
Ushering in the molecular millennium, the original draft of the Human Genome Project was completed in February 2001 through a multinational effort and has provided the architectural blueprints of essentially every gene in the human genome.1,2 The human genome embodies the total genetic information, or the deoxyribonucleic acid (DNA) content of human cells, and is dispersed among 46 units of tightly packaged linear double-stranded DNA called chromosomes (22 autosomal pairs and the 2 sex chromosomes X and Y).3–5 The 24 unique chromosomes are differentiated visually by chromosome-banding techniques (karyotype analysis) and are classified mainly according to their sizes. Each nucleated cell in a living organism normally has a complete and exact copy of the genome, which is largely made up of single-copy DNA with specific sets of DNA sequences represented only once per genome. The remainder of the genome consists of several classes of either perfectly repetitive or imperfectly repetitive DNA elements. The human genome contains nearly three billion base pairs of genetic information containing the molecular design for approximately 35,000 genes whose highly orchestrated expression renders us human.1,2 Through the mechanism of alternative splicing of the coding sequences within the genes, these approximately 35,000 genes are thought to produce more than 100,000 proteins.6
Basic Structure of DNA and the Gene
In 1953, Watson and Crick described the basic structure of DNA as a polymeric nucleic acid macromolecule comprising deoxyribonucleotides or “building blocks,” of which there are four types: (1) adenine (A), (2) guanine (G), (3) thymine (T), and (4) cytosine (C) (Figure 6-1).3–5 DNA is a double-stranded molecule made up of two anti-parallel complementary strands (sense and antisense strands) that are held together by noncovalent (loosely held) hydrogen bonds between complementary bases, where G and C always form base pairs and T and A always pair (see Figure 6-1). In the literature, typically only the DNA sequence of the sense strand (the strand that transcribes the genetic message in the form of messenger RNA [mRNA]) is provided, and the antisense sequence is inferred through these complementary base pairing rules such that if the sense strand reads AGCCGTA, the antisense strand would be TCGGCAT. DNA natively forms a double helix that resembles a right-handed spiral staircase. DNA elements that store genetic information in the form of a genetic code are called genes (Figure 6-2, A).
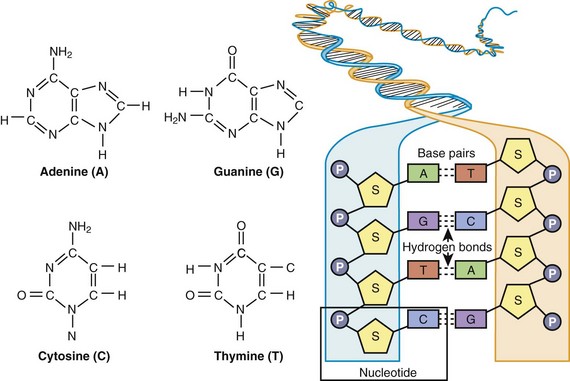
Figure 6-1 The chemical structure of adenine (A), guanine (G), cytosine (C), and thymine (T) and the general organization of DNA, illustrating complimentary base pairing via hydrogen bonds between C and G and between A and T. As defined by the box, a single nucleotide consists of a phosphate (P) group, deoxyribose sugar (S), and a base (A, C, G, or T).
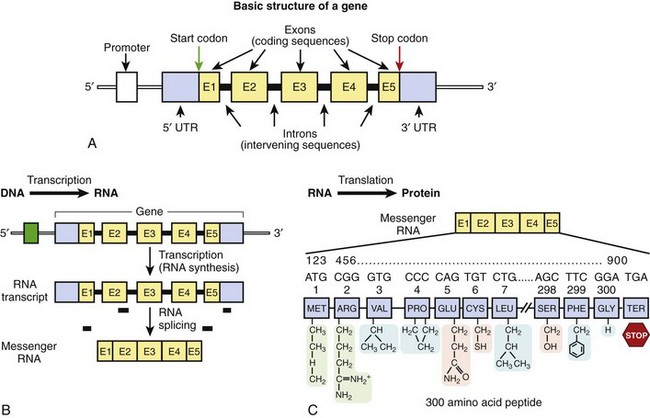
Figure 6-2 A, The basic structure of a gene consisting of DNA segments (exons) that encode for a protein product. Between the exons are intervening sequences called introns. At the 5’ end of the gene is a regulatory element called the promoter, which initiates transcription. At the 5’ and 3’ ends are “untranslated” regions that are considered parts of the first and last exons, respectively. These sequences are not a part of the genetic code but may contain additional regulatory elements. A start codon begins the translation of the genetic message, as encoded by the gene, and a stop codon terminates the message. B, Transcription. C, Translation. B and C depict the two-step process of the transfer of genetic information from DNA to RNA to protein.
Gene sequences account for approximately 30% of the genome; however, less than 2% of the genomic DNA is actually made up of protein-encoding sequences within genes called exons. Between the exons are intervening DNA sequences called introns, which are not a part of the genetic code but may host gene regulatory elements. The approximately 35,000 genes of the genome range in size from one of the smallest of human genes IGF2 (which contains 252 nucleotides and encodes insulin-like growth factor II) to the largest gene DMD (which consists of 2,220,223 nucleotides and encodes dystrophin). The DMD gene consists of over 2 million nucleotides, but only 0.5% of the gene (11,055 nucleotides spanning 79 exons) actually encodes for the dystrophin protein. Typically upstream (20 to 100 bp) from the first exon is a regulatory element called the promoter, which controls transcription of the hereditary message as determined by the gene sequence. Proteins known as transcription factors bind to specific sequences within the promoter region to initiate transcription of the genetic code. The first and last exons of the gene usually consist of an untranslated region (5′ and 3′ UTR, respectively) that is not a part of the genetic code but may host additional sequence elements that regulate gene expression.4
Transfer of the Genetic Code: The Central Dogma of Molecular Biology
DNA sequences in the form of genes contain an encrypted genetic message for the assembly of polypeptides or proteins that serve the biologic function of the cell. This inherited genetic information is transferred to a completed product (protein) through a two-step process.5 First, transcription, which is the process by which the genetic code is transcribed into mRNA, begins with the dissociation of the double-stranded DNA molecule and the formation of a newly synthesized complementary ribonucleic acid (RNA) molecule (Figure 6-2, B). Of note, instead of thymine (T), the nucleotide uracil (U) is in its place on the newly transcribed RNA strand; like thymine, uracil pairs with adenine. The initial mRNA molecule (pre-mRNA) matures into a transferable genetic message by undergoing RNA splicing to expunge the noncoding intronic sequences from the transcript. The vast majority of introns begin with the di-nucleotides GT and end with the di-nucleotides AG. These highly conserved splicing recognition sequences at the beginning and end of the exon-intron and intron-exon boundaries are referred to as the splice donor sites and splice acceptor sites, respectively. These nucleotides allow the RNA splicing apparatus to know precisely where to cleave the sequence in order to excise the noncoding regions (introns) and bring the coding sequences (exons) together. Normal alternative splicing provides the inclusion or exclusion of specific exonic sequences from the mature mRNA transcript to potentially produce several partially unique gene products (proteins) from a single gene that may have unique biologic functions, tissue specificity, or cellular locations. If normal splice recognition sites are disrupted, splicing errors may occur and result in abnormal protein product formation and consequently create a pathogenic substrate for disease. While all cells of the human body, except red blood cells, contain a copy of the genome, not all genes are expressed in all cells. While some genes are ubiquitously expressed, others have exclusive tissue specificity.
The second process, translation, involves the decoding of the mRNA-encrypted message and the assembly of the intended polypeptide (protein) that will serve a biologic role (Figure 6-2, C). Polypeptides are polymers of linear repeating units called amino acids. The assembly of a polypeptide or protein is directed by a triplet genetic code, or codon (three consecutive bases); 64 codons encode for 20 distinct amino acids or the termination of protein assembly. One codon, AUG (ATG on DNA) encodes for the amino acid methionine and is always the first codon (start codon) to start the message and signifies the beginning of the open reading frame (ORF) of the mRNA. Each codon in the linear mRNA is decoded sequentially to give a specific sequence of amino acids that are covalently linked through peptide bonds and ultimately make up a protein. Three codons, UAA, UAG, and UGA, serve as termination codons that stop the linearization of the peptide and signal a release of the finished product. The genetic code is said to be “degenerate” in that specific amino acids may be encoded by more than one codon. For example, when varying the nucleotide in the third position of a codon, often the message does not become altered (the codons GUU, GUC, GUA, and GUG all encode for the amino acid valine).
The accepted nomenclature for naming and numbering nucleotides and codons typically uses the DNA sense strand of the gene and begins with the A of the start codon (ATG) representing nucleotide 1 and ATG as codon 1. Usually, only consecutive nucleotides within the coding region of the gene are numbered. Intronic nucleotides are typically numbered relative to either the first or last nucleotide in the exon preceding or following the intron. For example, the LQT2-associated KCNH2 splice error mutation L799sp (exon 9, nucleotide substitution: 2398 +5 G > T), results from a G-to-T substitution in the intron, five nucleotides from exon 9, where nucleotide 2398 is the last nucleotide in the ninth exon. This substitution results in a splicing error following the last codon of the exon [codon 799 encoding for leucine (L)].7
Non–protein-coding genes are transcribed as well. MicroRNAs (miRNAs) are small ~22 nucleotide-long RNAs that function to inhibit gene expression of targeted genes by binding in a partially complementary fashion to miRNA recognition sequences within the 3′ UTR of target mRNA transcripts and negatively regulate protein-encoding gene mRNA stability or translation into protein.8–10 Each miRNA is thought to regulate the expression of hundreds of target genes at the post-transcriptional mRNA level. To date, hundreds of human miRNAs have been described, three (miR-1, miR-133, and miR-208) of which are abundant in the heart and serve as key regulators of heart development, contraction, and conduction.8
Modes of Inheritance: Genetics of Disease
On average, two unrelated individuals share 99.5% of their approximately three billion nucleotide genomic DNA sequence, and yet their genomic DNA sequences may vary at millions of single nucleotides or small sections of DNA nucleotides dispersed throughout their genomes.2,11 It is this inherited variation in the genome that is the basis of human and medical genetics. Reciprocal forms of genetic information at a specific locus (location) along the genome are called alleles.3 An allele can represent a segment of DNA or even a single nucleotide. The normal form of genetic information is often considered the wild-type or normal allele, and the allele at variance from the normal is often referred to as the mutant allele.
These normal variations at specific loci in the DNA sequence are called polymorphisms. Some polymorphisms are very common, and others represent rare genetic variants. In medical genetics, a disease-causing mutation refers to a DNA sequence variation that embodies an abnormal allele and is not found in the normal healthy population but subsists only in the diseased population and produces a functionally abnormal product. An individual is said to be homozygous when he or she has a pair of identical alleles, one paternal (from father) and one maternal (from mother). When the alleles are different, then that individual is said to be heterozygous for that specific allele. The term genotype refers to a person’s genetic or DNA sequence composition at a particular locus or at a combined body of loci, and the term phenotype refers to a person’s observed clinical expression of disease in terms of a morphologic, biochemical, or molecular trait.5
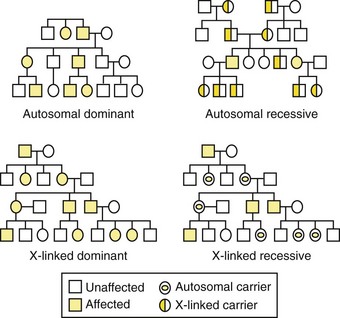
Genetic disorders are described by their patterns of familial transmission (Figure 6-3). The four basic modes of inheritance are (1) autosomal dominant, (2) autosomal recessive, (3) X-linked dominant, and (4) X-linked recessive.3 These modes, or patterns, of inheritance are based mostly on the type of chromosome (autosome or X-chromosome) the gene is located on and whether the phenotype is expressed only when both maternal-derived and paternal-derived chromosomes host an abnormal allele (recessive) or if the phenotype can be expressed even when just one chromosome of the pair (maternal or paternal) harbors the mutant allele (dominant).
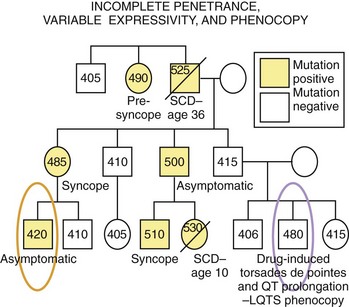
In many genetic disorders, the abnormal phenotype can be clearly distinguished from the normal one. However, in certain disorders, the abnormal phenotype is completely absent in some individuals (asymptomatic, with no discerning clinical markers) harboring the disease-causing mutation, while some others show significant variations in the expression of the phenotype in terms of clinical severity, age at onset, and response to therapy. Penetrance is the probability that an abnormal phenotype, as a result of a mutant gene, will have any expression at all. When the frequency of phenotypic expression is less than 100%, the gene is said to show reduced or incomplete penetrance (Figure 6-4). Expressivity refers to the level of expression of the abnormal phenotype, and when the manifestations of the phenotype in individuals who have the same genotype are diverse, the phenotype is said to exhibit variable expressivity (see Figure 6-4). A phenocopy represents an individual who displays the clinical characteristics of a genetically controlled trait but whose observed phenotype is caused by environmental factors rather than determined by his or her genotype (see Figure 6-4). For example, an individual experiencing drug-induced torsades de pointes, a prolonged QT interval on ECG, or both may represent a phenocopy of LQTS. Reduced penetrance, variable expressivity, and observed phenocopies create significant challenges for the appropriate diagnosis, pedigree interpretation, and risk stratification of some genetic disorders, particularly those involving electrical disorders of the heart.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


