Chapter 8 Disorders of Intracellular Transport and Intercellular Conduction
Disorders of Intercellular Trafficking
Formation of Protein Subunits
As membrane proteins are formed in the ER, they undergo folding to their correct functional conformation. Occasionally, this process does not work correctly. In order to ensure that nonfunctional proteins are not targeted to the myocyte membrane, a process called unfolded protein response occurs. ER-associated degradation (ERAD) is responsible for the elimination and control of the buildup of aberrant proteins, preventing the aggregation of toxic nonfunctional proteins.1 These misfolded proteins are recognized by chaperones such as Hsp70/Hsp40, which interact directly with the misfolded protein and aid in its translocation into the cytoplasm for degradation by the proteasome. Evidence is now beginning to accumulate that the ERAD pathway participates not only in the removal of proteins that are not folded correctly but also in the rapid degradation of excess proteins. Thus, a protein is made from RNA in larger quantities than needed under normal conditions but is degraded via the ERAD pathway prior to insertion into cell membranes. When a cell needs a rapid increase of a particular protein, the ERAD system is turned off and more of the protein is trafficked to the cell membrane for use. An example of this is the adenosine-5′-triphosphate (ATP)–sensitive potassium (K[ATP]) channel, whose biogenesis and surface expression are controlled by ERAD. This is thought to be a mechanism for rapid increase in the availability of K(ATP) channels during cellular stress.2
Genetic Disorders of Trafficking
Normal trafficking of membrane proteins is exquisitely dependent on having particular amino acid sequences within the protein sequence. These sequences allow for the interaction of the channel protein with molecular motors, cytoskeletal elements, and scaffolding proteins, which are all important for protein movements throughout the cell. Changes in the nucleotide sequence of DNA will translate into RNA missense errors, which, in turn, lead to the production of incorrect amino acid sequences within the proteins. Thus, errors within the genome may produce proteins that are missing part or all of the appropriate trafficking sequences, thus causing genetic trafficking disorders. Since the normal cardiac rhythm is dependent on ion channels and gap junctions, abnormal ion channel and gap junction function underlie a number of cardiac rhythm disturbances. Alteration in individual channel gating function was thought to cause much of the loss of function of mutated channels, but analysis of many of the proarrhythmic mutations indicates that the true cause is the loss of trafficking to the membrane surface. For example, an alteration in the trafficking of ion channels to the cell’s surface has been shown to occur in some forms of atrial fibrillation (AF), long QT syndrome (LQTS) types 1 and 2, Brugada syndrome, and Anderson syndrome (Table 8-1).
One of the more common forms of changes within protein sequences occurs when a coding change produces a protein with alternative amino acids at a given position within the protein. This shift, known as polymorphism, has two possible amino acids at particular loci within a protein but leaves the remaining amino acids in the correct location within the protein sequence. In many cases, no phenotypic change associated with polymorphisms occurs, but in rare cases, the changes at a single site can lead to the production of a protein that is nonfunctional. For example, in SCN5A, a mutation at amino acid 1053 from glutamic acid (E) to lysine (K) leads to a trafficking disorder of the sodium (Na+) channel. Written as I1053K, this mutation prevents the SCN5A protein from localizing to cell membranes by interrupting the ability of the SCN5A protein to interact with the scaffolding protein ankyrin G, which normally maintains the protein subunit at the cell membranes (see Chapter 2 and Table 8-1). This simple substitution of a single amino acid thus causes the Brugada syndrome phenotype.
Frame shift mutations may also occur. In these mutations, a coding error causes the initial RNA triplet to have either an extra nucleotide or a lost nucleotide, shifting the reading frame in a manner that causes a novel amino acid to be produced at that site. The produced protein will then have a scrambled or “nonsense” sequence of amino acids. These errors are very severe and often lead to proteins that are trafficking deficient; in addition, if the trafficking deficiency is rescued pharmacologically, these proteins are unable to form a channel with normal function. One example of this is the LQTS type 1 (LQT1) mutation that results from a frame shift at position 178 in the α-subunit of the channel that underlies the slow component of the delayed rectifier current, IK. In this case, the alanine normally found at position 178 is lost, which leads to the formation of an abnormal amino acid sequence; this sequence ends 105 amino acids later when, by chance, the combination of nucleotides forms a stop codon and the protein translation is terminated. This type of mutation, written as A178fs/105 in this case, causes the formation of a truncated form of the protein that is trafficking deficient and leads to the LQT1 phenotype (see Table 8-1).
Sodium Channels
The primary phenotype found in patients with mutations that cause loss of trafficking of sodium (Na+) channels to the cell membranes is Brugada syndrome. This phenotype is associated with high risk for sudden cardiac death. Voltage-gated Na+ channels are responsible for the rapid upstroke of the cardiac action potential and persistence of some Na+ current after rapid depolarization participates in the early phase of repolarization. Conduction velocity is, in part, dependent on both the amplitude and the rate of activation of these channels, and repolarization is normally slowed by the persistence of Na+ inward current during the early plateau phase. Therefore, decreased Na+ current leads to a shortened action potential duration. This exaggerates the normal heterogeneity of the outward current found in the heart. This heterogeneity is usually masked by the inward Na+ current leading to a voltage gradient across the ventricular wall, which is evident on the ECG as the classic Brugada’s ST-segment elevations in leads V1 through V3.3 Brugada syndrome can also result from mutations in the channel proteins that cause changes in channel function rather than a trafficking defect. Thus, loss of properly functioning Na+ channels at the cell membranes—either because of direct Na+ channel mutations or through loss of interaction of the Na+ channel with trafficking or scaffolding partners—leads to reduced Na+ current, increased heterogeneity in repolarization, and the subsequent production of re-entrant arrhythmias leading to sudden cardiac death.
Potassium Channels
Potassium (K+) channel mutations that decrease total channel expression at cell membranes are an important cause of LQTS types 1 and 2 in addition to other mutations that cause loss of function of channels that are trafficked to the membranes. The primary current affected in LQTS is the delayed outward rectifier K+ current, IK. Depending on the channel in which a mutation resides, the subtypes of K+ current affected (IKr or IKs) may vary, but the overall effect that manifests as LQTS is a loss of outward K+ current. This loss leads to a delay in ventricular cell repolarization and the duration of the QT interval. As with Brugada syndrome, this leads to an increase in transmural repolarization gradients. The mechanism for ventricular tachycardia (torsades de pointes, TdP) associated with LQTS has been postulated to be the occurrence of early after-depolarizations (EADs) and triggered activity caused by the action potential prolongation, which leads to re-entry facilitated by the heterogeneities of repolarization.3
An additional K+ channel trafficking mutation associated with cardiac arrhythmias and sudden cardiac death is Andersen-Tawil syndrome (see Table 8-1). This pleiotropic disorder is caused by a primary mutation in the gene KCNJ2. This gene codes for the Kir2.1 channel, which underlies the inwardly rectifying K+ channel. As with other K+ channel trafficking mutations, the primary ECG manifestation is a longer than normal QT interval, a dispersion of repolarization, and an increase in the propensity for ventricular arrhythmias likely triggered by EADs. While Andersen-Tawil syndrome has electrocardiographic similarities to LQTS, the pleiotropic nature of the disorder (periodic paralysis and dysmorphic features) helps distinguish it from other LQTSs.
Gap Junction Channels
Although deficiencies in gap junction function are associated with many different cardiac arrhythmias (see below), to date, the only arrhythmia associated with trafficking deficient mutations of gap junction proteins is AF. In some patients with AF, a subset of trafficking deficient mutations is found in connexin40 (Cx40), a primary gap junction protein in the atria.4 Interestingly, these mutations also cause a dominant negative effect on the trafficking of connexin43 (Cx43), the other connexin isoform found in the atria, leading to very low levels of cell–cell coupling between the atrial myocytes. The mechanism for this transdominant effect is not clear, but the overall effect is loss of cell–cell coupling, which leads to slowed conduction and a propensity for the formation of re-entrant arrhythmias. Trafficking mutations in Cx43, which is the major gap junction protein in the ventricular myocardium, have not been found to be associated with ventricular arrhythmias. This is primarily because Cx43 is vital for normal cardiac development and function; thus, trafficking mutations would likely cause embryonic lethality.
Intercellular Communication
The ability for the heart to pump efficiently is dependent on the syncytial nature of the myocardium. Coordination of the contraction is maintained by passage of the electrical current through gap junctions, which are specialized membrane channels.5 Gap junctions are formed from half, or hemi-, channels inserted into the plasma membranes of individual myocytes. These hemi-channels (connexons) localize to the intercalated disc and meet head-to-head across the extracellular space with a connexon from an adjacent cell. This forms a longitudinally oriented conduit, which rapidly spreads excitation throughout the heart (Figure 8-1). The importance of gap junctions in maintaining conduction is underscored by the fact that their loss has been associated with slowed conduction and the formation of re-entrant arrhythmias.
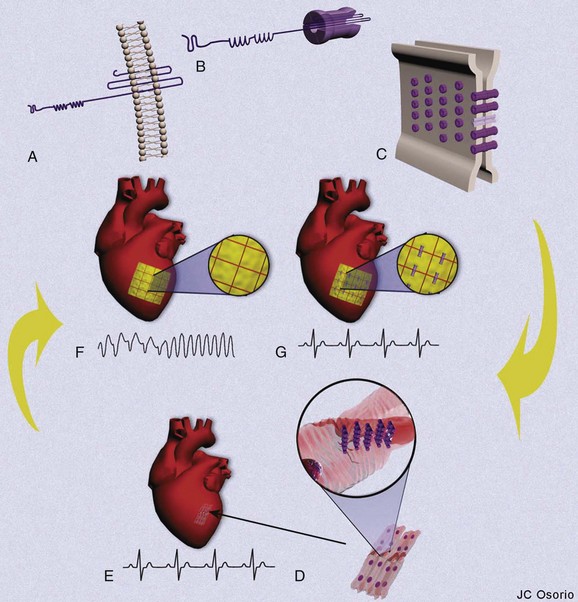
Connexons are formed from the oligomerization of proteins from the connexin family in the Golgi apparatus. These proteins form four transmembrane domain-spanning units (see Figure 8-1). Overall, there are 21 isoforms of connexin in the human genome, five of which are found in the heart (Table 8-2).5 The most abundant connexin in the heart is Cx43, which localizes to both the atrial and the ventricular myocytes. The second most abundant connexin is Cx40, which is found in the atrial myocytes, largely co-localizing with Cx43 but is also a predominant isoform within the nodes of the heart. It is found more sparsely within the specialized conduction system in conjunction with connexin45 (Cx45). Low levels of this connexin have been reported in both the atria and the ventricles, although the physiological relevance Cx45 in these regions is unclear. The nodes of the human heart also contain connexin31.9 (Cx31.9), a low-conductance gap junction channel. Connexin37 (Cx37) occurs in the endothelial lining of the cardiac vasculature in conjunction with Cx43.5
Table 8-2 Connexins in the Heart
| Cx31.9 | |
| Cx37 | Endothelial cells of vessels |
| Cx40 | |
| Cx43 | |
| Cx45 |
Connexins follow the general pattern of membrane protein trafficking outlined above, with some interesting additions. The threading of the connexin protein into the membrane occurs, as with all membrane proteins, as it is being formed in the ER, but then the six connexins that form the connexon oligomerize, most likely beginning this process in the ER but finalizing the connexon formation within the Golgi apparatus.6 Thus, the protein subunits form the channel prior to insertion into the membrane of the cell and must be regulated to stay closed to ensure that the intracellular compartments do not exchange components.
Once the connexons are formed, they traffic out to the plasma membrane within vesicles and in some cases, such as Cx43, within caveolae with the aid of microtubules.7,8 For nonpolarized cells such as epithelial cells, a nondirected movement of connexons occurs, when connexons are trafficked outward to any region of the cell membrane. Polarized cells such as cardiomyocytes show directionality in the trafficking, with connexons being shuttled to the membrane domains in which they will reside and function. In the cardiac myocyte, this is the intercalated disc. From here, connexons move through the lipid bilayer and accumulate at the edges of gap junctional plaques, increasing the size of the plaque. As the protein is added, older protein that is designated to be removed from the plaque coalesces in the center of the plaque, and removal occurs from there.9 This turnover is rapid, occurring within a few hours, giving an exquisite regulatory control to the level of junctional coupling between cells.
Abnormalities in Intercellular Communication Causing Cardiac Arrhythmias
Changes in Gap Junction Conductance Without Changes in Connexin Amount or Location
The gap-junctional membrane provides low-resistance pathways for current flow between myocardial cells as well as for the passage of small molecules (up to 1 kDa). The permeability of a gap junction to the flow of ions that carry current (i.e., junctional conductance) is determined by the number of junctional channels, the proportion of the channels that are in the open state, and the permeability (conductance) of the open channel. The permeability of the gap junction channel in the open state, or unitary conductance, is determined by the particular connexin isoform or combination of isoforms that form the channel.5
The conductance of gap junctional channels may change under pathophysiological conditions without any change in the quantity or location of the connexin protein, through a change in the average conductance of gap junction channels. An important cause of reduction in Cx43 gap junction coupling that can occur in the ventricular ischemic arrhythmogenic substrate is low intracellular pH (below 6.5), an effect that can be facilitated by high intracellular Ca2+. Effects of Ca2+ and pH are caused by a change in the open probability of the gap junction channel rather than by single-channel conductance. pH becomes an important direct influence at the low pH (<6.5) associated with hypoxia and ischemia by directly closing the channels, but ischemia and hypoxia may also lead to changes in connexon configuration that contribute to decreased gap junction conductance.6
Changes in the phosphorylation state of connexins during remodeling can change gap junction channel function.6 For example, Cx43 is normally phosphorylated at multiple serine residues. Phosphorylation plays a number of roles. It might be necessary for maintaining hemi-channels in their closed state until docking occurs in the membrane of the intercalated disc. It also appears to be necessary for the normal opening and closing of gap junction channels. Changes in the phosphorylation state during acute ischemia can decrease the open probability of gap junction channels, probably prior to the change in Cx43 quantity that occurs following approximately 15 minutes of ischemia.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


