Coronary Thrombosis: Local and Systemic Factors: Introduction
The formation of an acute thrombus on a ruptured coronary atherosclerotic lesion, obstructing coronary blood flow and reducing the oxygen supply to the myocardium, leads to the onset of acute coronary syndromes (ACS). These thrombotic episodes largely occur in response to atherosclerotic lesions that have progressed to a high-risk inflammatory or prothrombotic stage. Although they are distinct from one another, the atherosclerotic and thrombotic processes appear to be closely interrelated, causing ACS through a complex, multifactorial process called atherothrombosis. ACS represents a spectrum of ischemic myocardial events that share similar pathophysiology; they include unstable angina/non–ST-segment elevation myocardial infarction (UA/NSTEMI), ST-segment elevation myocardial infarction (STEMI), and sudden cardiac death.
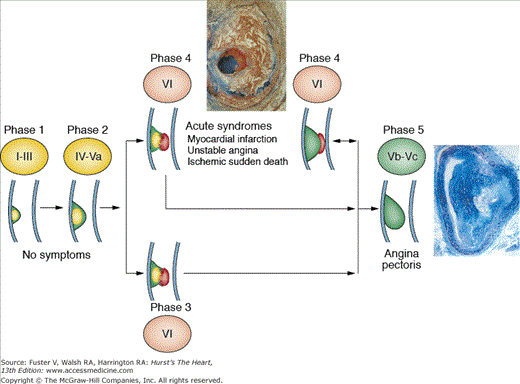
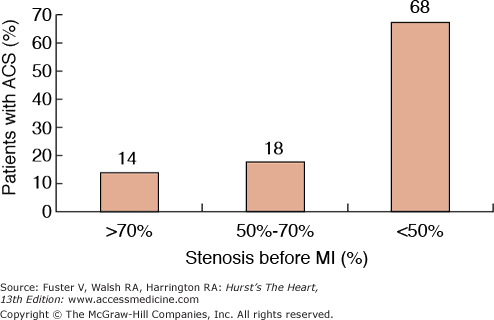
Atherosclerosis is a systemic disease involving the intima of large and medium-sized arteries, including the aorta, carotids, coronaries, and peripheral arteries, that is characterized by intimal thickening caused by the accumulation of cells and lipids (Fig. 53–1).1 Lipid accumulation results from an imbalance between the mechanisms responsible for the influx and efflux of lipids into the arterial wall.2 Secondary changes may occur in the underlying media and adventitia, particularly in advanced disease stages. The early atherosclerotic lesions might progress without compromising the lumen because of compensatory vascular enlargement (Glagovian remodeling).3 Importantly, the culprit lesions leading to ACS are usually mildly stenotic and therefore barely detected by angiography (Fig. 53–2).4 These high-risk, rupture-prone lesions usually have a large lipid core, a thin fibrous cap, and a high density of inflammatory cells (particularly at the shoulder region, where disruptions most often occur).
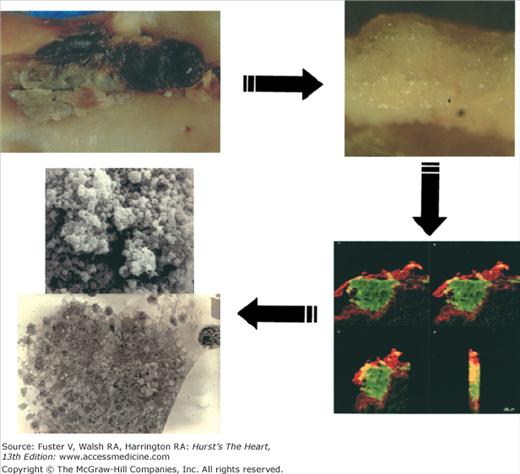
Recent evidence has highlighted the importance of lesion neovascularization and blood extravasation in plaque destabilization and plaque growth.5-7 Leaky vasa vasorum with the subsequent red blood cell extravasation has been postulated as a major source for intraplaque cholesterol deposition. This change in composition, characterized by increased extracellular cholesterol within the lipid core and excessive macrophage infiltration, increases the vulnerability of the atherosclerotic lesions. In fact, postmortem studies have shown a strong correlation between macrophage infiltration and increased vasa vasorum in human atherosclerotic lesions. Preexisting vasa vasorum in the adventitia are thought to spread into the intima, prompting intimal neovascularization.6 Elimination of neovascularization for plaque stabilization has been postulated as a novel target to treat patients with high-risk plaques.8 Inflammation is another important process that affects plaque progression, vulnerability, and subsequent thrombus formation. Inflammation could be considered as the link between atherosclerosis and thrombosis. In fact, the relationship of inflammation and atherothrombosis could represent different faces of the same disease. Under “healthy” circumstances, the normally functional endothelial monolayer creates an antiatherogenic environment protecting against atherogenesis. This protection is achieved by releasing a series of antithrombotic and vasoactive substances (eg, nitrous oxide [NO], prostacyclin [PGI2], tissue plasminogen activator [tPA]). The initial pathological manifestation of arthrosclerosis is a dysfunctional endothelium. The dysfunctional endothelium, characterized by a reduced NO and PGI2 synthesis, facilitates the permeability of circulating lipids into the subendothelial space and, by exposing adhesive proteins of the selectin family, it facilitates the “homing’ of the circulating monocytes on the endothelium. Vascular cell adhesion molecule-1 (VCAM-1) is implicated in early adhesion of mononuclear leukocytes to arterial endothelium at sites of atheroma initiation.9 In addition to VCAM-1, P- and E-selectin also seem to contribute to leukocyte recruitment to the nascent atheroma plaque.10,11 There are many other “inflammatory” molecules implicated in atherogenesis, including monocyte chemotactic protein-1(MCP-1), T lymphocytes, interleukin-1 (IL-1), and interleukin-6 (IL-6).
Platelets also play an important role in the inflammatory environment. Although formerly platelets were suggested to be passive players because they are organelles lacking nucleus, currently we know that they are very active, being important not only in thrombosis itself but also in perpetuating the inflammatory environment. Platelets secrete various vasoactive chemokines and cytokines, including CD40L, thrombospondin, platelet-activating factor, RANTES (regulated upon activation, normal T-cell expressed and secreted), epithelial-derived neutrophil-activating peptide 78 (ENA-78), macrophage inflammatory protein (MIP), chemokine (C-X-C motif) ligand 4 (CXCL4), with autocrine and paracrine effects. Also very important is the notion that free cholesterol, a major component of atherosclerotic plaques, is one the most inflammatory substances triggering the recruitment of more macrophages. Therefore, inflammation plays a dual role on atherothrombosis both at the vascular and circulating levels.12
A reliable, noninvasive imaging tool able to detect early atherosclerotic disease and characterize lesion composition would be clinically advantageous. Although magnetic resonance imaging (MRI) has been widely tested for plaque composition analysis,13 other imaging techniques, such as computed tomography (CT)14,15 and radionuclide imaging (positron emission tomography [PET])16 have recently been used for this enterprise (see Chaps. 24 and 25). The reliable noninvasive plaque characterization and eventually the vulnerable plaque identification would improve our understanding of the pathophysiologic mechanisms of atherothrombosis and help in the risk stratification of the patients to select the appropriate approach.17
Growing thrombi on atherosclerotic vessels may occlude the lumen locally or embolize and be washed away by the blood flow to occlude distal vessels. However, thrombi may be physiologically or spontaneously lysed by mechanisms that block thrombus propagation. Thrombus size, location, and composition are regulated by local fluid dynamic conditions (mechanical effects), the thrombogenicity of exposed substrate (local molecular effects), the relative concentration of fluid phase and cellular blood components (local cellular effects), and the efficiency of the physiologic mechanisms of control of the system, mainly fibrinolysis.18 Similarly, the size and stability of the thrombus are major modulators of the severity of the ACS.
Cellular and Molecular Mechanisms in Thrombus Formation
Although several decades ago the endothelium was viewed as a simple barrier separating the fluid phase of the blood from the highly thrombogenic smooth muscle vascular wall, today we know that the endothelium is a critical player in the maintenance of the normal function of the arterial bed.19 The endothelium (endothelial cells) constantly secretes substances (eg, hormones, growth factors) into the vascular lumen to maintain vascular tone and to avoid abnormal platelet adhesion or activation and clot formation. When the endothelium is damaged and cannot perform this crucial task, it is dysfunctional. Endothelial dysfunction as well as a discontinuity of the endothelial integrity triggers a series of biochemical and molecular reactions aimed at preventing excessive blood loss and repairing the vessel wall. Vasoconstriction and platelet adhesion at the site of injury combine to form a hemostatic aggregate as the first step in vessel wall repair and the hemostasis. A few platelets may interact with injured and dysfunctional endothelium and release growth factors that stimulate intimal hyperplasia.9 Several layers of platelets may be deposited on the lesion with mild injury and may or may not evolve to become a mural thrombus. The release of platelet growth factors may contribute significantly to accelerated intimal hyperplasia, as occurs in the coronary vein grafts within the first postoperative year. With severe injury and exposure of components of deeper layers of the vessel, as in spontaneous plaque rupture and in angioplasty, marked platelet aggregation with mural thrombus formation follows. Vascular injury of this magnitude also stimulates thrombin formation through both the intrinsic (surface-activated) and extrinsic (tissue factor [TF]–dependent) coagulation pathways (Fig. 53–3).
Plaque rupture facilitates the interaction of inner plaque components with the circulating blood. Among these components, TF exhibits a potent activating effect on platelets and coagulation. There is now understanding of the biochemical events involved in platelet activation.19,20 At the site of vascular lesions, circulating von Willebrand factor (vWF) binds to the exposed collagen which subsequently binds to the glycoprotein (GP) Ib/IX receptor on the platelet membrane.21-23 Under pathological conditions and in response to changes in shear stress, vWF can be secreted from the storage organelles in platelets or endothelial cells, reinforcing the activation process. Although GP Ib/IX–vWF interaction is enough to promote binding of platelets to subendothelium, it is highly transient, resulting in rapid dislocation of platelet to the site of injury. GPVI binding to matrix collagen has slower binding kinetics, but when initiated, it promotes a firm adhesion of platelet to the vessel surface.24 Figure 53–4 shows mechanisms and agonists involved in platelet adhesion, activation, and aggregation.25 Finally, both GPIb/IX and GPVI also regulate platelet–leukocyte adhesion and thereby are implicated in other vascular process, such as inflammation and atherosclerosis.26-28 Exposed matrix from the vessel wall and thrombin generated by activation of the coagulation cascade, as well as epinephrine and adenosine diphosphate (ADP), are powerful platelet agonists. Each agonist stimulates the discharge of calcium and promotes the subsequent release of its granular content. Platelet-related ADP and 5-hydroxytryptamine (5-HT) stimulate adjacent platelets, further enhancing the process of platelet aggregation. Arachidonate, which is released from the platelet membrane by the stimulatory effect of collagen, thrombin, ADP, and 5-HT, promotes the synthesis of thromboxane A2 (TXA2) by the sequential effects of cyclooxygenase (COX) and thromboxane synthetase. TXA2 not only promotes further platelet aggregation but is also a potent vasoconstrictor (Fig. 53–5; see also Fig. 53–4). The initial recognition of damaged vessel wall by platelets involves (1) adhesion, activation, and adherence to recognition sites on the thromboactive substrate (extracellular matrix [ECM] proteins such as vWF, collagen, fibronectin, vitronectin, and laminin); (2) spreading of the platelet on the surface; and (3) aggregation of platelets to form a platelet plug or white thrombus. Figure 53–6 shows the processes implicated in the formation of a thrombus upon a plaque disruption.25 The efficiency of platelet recruitment depends on the underlying substrate and local geometry (local factors). A final step involving the recruitment of other blood cells also occurs; erythrocytes, neutrophils, and occasionally monocytes are found on evolving mixed thrombus.
Figure 53–4.
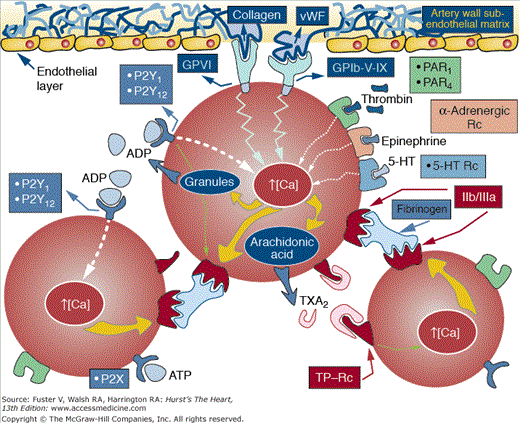
Mechanisms and agonists involved in platelet adhesion, activation, and aggregation. ATP, adenosine triphosphate; 5-HT, 5-hydroxytryptamine; GP, glycoprotein; PAR, protease-activated receptor; TP, thromboxane receptor, TXA, thromboxane; vWF, von Willebrand factor. Reprinted from Ibanez, Vilahur, Badimon.25 Under permission of the copyright holder: Oxford University Press.
Figure 53–5.

Signal transduction mechanisms of platelet activation and aggregation. AA, arachidonic acid; ADP, adenosine diphosphate AMP, adenosine 3—5—-cyclic monophosphate; ATP, adenosine triphosphate; DG, diacylglycerol; Gs, Gi, Gp, Gq, guanine nucleotide-binding regulatory proteins; 5HT, 5-hydroxytryptamine; IP3, inositol 1,4,5-triphosphate; PGI2, prostacyclin; PIP2, phosphoinositol diphosphate; PKCi and PKCa, protein kinase C, inactivated and activated; PLA2, phospholipase A2; PLC, phospholipase C; IIb/IIIa, receptor glycoprotein for adhesive protein ligands (mainly fibrinogen and vWF); TXA2, thromboxane A2.
Figure 53–6.
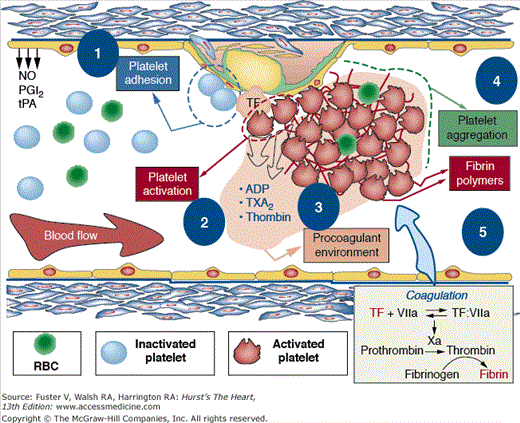
Mechanism involved in thrombus formation. Healthy endothelium (left) presents antithrombotic properties because it is able to release vascular protective substances such as nitric oxide (NO), prostacyclin (PGI2), tissue plasminogen activator (tPA), and tissue factor pathway inhibitor (TFPi). On the contrary, dysfunctional endothelium (right) not only favors platelet adhesion, activation, and aggregation but also promotes vascular lipid deposition, macrophage migration, and TF expression (activation of the coagulation cascade). After platelet adhesion, activation is characterized by platelet shape change. Activated platelets secrete different agonists, prompting activation of circulating platelets and a procoagulant environment. This prothrombotic milieu favors thrombus formation and the subsequent clinical manifestations. ADP, adenosine diphosphate; RBC, red blood cell; TXA2, thromboxane A2. Reprinted from Ibanez et al.25 Under permission of the copyright holder: Oxford University Press.
Platelet function depends on the adhesive interaction of several compounds. Most of the GPs on the platelet membrane surface are receptors for adhesive proteins. Many of these receptors have been identified, cloned, sequenced, and classified within large gene families that mediate a variety of cellular interactions29,30 (Table 53–1). The most abundant is the integrin family, which includes GP IIb/IIIa, GP Ic/IIa, the fibronectin receptor, and the vitronectin receptor, in decreasing order of magnitude. Another gene family present in the platelet membrane glycocalyx is the leucine-rich GP family represented by the GP Ib/IX complex, receptor for vWF, on unstimulated platelets that mediates adhesion to subendothelium and GP V. Other genes include the selectins (such as GMP-140) and the immunoglobulin domain protein (human leukocyte antigen [HLA] class I antigen and platelet/endothelial cell adhesion molecule 1 [PECAM-1]). Unrelated to any other gene family is GP IV (IIIa)29 (see Table 53–1).
| Glycoprotein Receptor | Function | Ligand |
|---|---|---|
| GP IIb/IIIa | Aggregation, adhesion at high shear rate | Fg, vWF, Fn, Ts, Vn |
| Receptor Vn | Adhesion | Vn, vWF, Fn, Fg, Ts |
| GP Ia/IIa | Adhesion | C |
| GP Ic/IIa | Adhesion | Fn |
| GP lcN/lla | Adhesion | Ln |
| GP Ib/IX | Adhesion | vWF, T |
| GP V | Unknown | Substrate T |
| GP IV (GP IIIb) | Adhesion | Ts, C |
| GMP-140 (PADGEM) | Interaction with leukocytes | Unknown |
| PECAM-1 (GP IIa) | Unknown | Unknown |
Randomly distributed on the surface of resting platelets are about 50,000 molecules of GP IIb/IIIa. The complex is composed of one molecule of GP IIb (disulfide-linked large and light chains) and one of GP IIIa (single polypeptide chain). It is a Ca2+-dependent heterodimer, noncovalently associated on the platelet membrane.31 Calcium is required for maintenance of the complex and for binding of adhesive proteins.32,33 On activated platelets, the GP IIb/IIIa is a receptor for fibrinogen, fibronectin, vWF, vitronectin, and thrombospondin.
The GP Ib/IX complex consists of two disulfide-linked subunits (GP Ibα and GP Ibβ) tightly (not covalently) complexed with GP IX in a 1:1 heterodimer. GP Ibβ and GP IX are transmembrane GPs and form the larger globular domain. The elongated, protruding part of the receptor corresponds to GP Ibα. The major role of GP Ib/IX is to bind immobilized vWF on the exposed vascular subendothelium and initiate adhesion of platelets. GP Ib does not bind soluble vWF in plasma; apparently, it undergoes a conformational change upon binding to the ECM and then exposes a recognition sequence for GP Ib/IX. The cytoplasmic domain of GP Ib/IX has a major function in linking the plasma membrane to the intracellular actin filaments of the cytoskeleton and functions to stabilize the membrane and to maintain the platelet shape.34,35
Thrombin plays an important role in the pathogenesis of arterial thrombosis. It is one of the most potent known agonists for platelet activation and recruitment. In addition, thrombin is critical in the maintenance of the fibrin mesh. The thrombin receptor has 425 amino acids with seven transmembrane domains and a large NH2-terminal extracellular extension that is cleaved by thrombin to produce a “tethered” ligand that activates the receptor to initiate signal transduction.36,37 Thrombin is a critical enzyme in early thrombus formation, cleaving fibrinopeptides A and B from fibrinogen to yield insoluble fibrin, which effectively anchors the evolving thrombus. Both free and fibrin-bound thrombin are able to convert fibrinogen to fibrin, allowing propagation of thrombus at the site of injury.
Therefore, platelet activation triggers intracellular signaling and expression of platelet membrane receptors for adhesion and initiation of cell contractile processes that induce shape change and secretion of the granular contents. The expression of the integrin IIb/IIIa (αIIbβ3) receptors for adhesive GP ligands (mainly fibrinogen and vWF) in the circulation initiates platelet-to-platelet interaction. The process is perpetuated by the arrival of platelets from the circulation. Most of the GPs in the platelet membrane surface are receptors for adhesive proteins or mediate cellular interactions. vWF has been shown to bind to platelet membrane GPs in both adhesion (platelet–substrate interaction) and aggregation (platelet–platelet interaction), leading to thrombus formation, as seen in perfusion studies conducted at high shear rates.38-41 Ligand binding to the different membrane receptors triggers platelet activation with different relative potencies. Great interest in the platelet ADP receptors (P2Y, P2X) has recently been generated because of available pharmacologic inhibitors. The P2Y1 receptor is responsible for inositol trisphosphate formation through activation of phospholipase C, leading to transient increase in the concentration of intracellular calcium, platelet shape change, and weak transient platelet aggregation.42-44 Pharmacologic data have revealed an essential role for the P2Y1 receptor in the initiation of platelet ADP-induced activation, TXA2 generation, and platelet activation in response to other agonists.45 The P2Y12 receptor is responsible for completion of the platelet aggregation response to ADP.46 There are several signaling molecules downstream of P2Y12 activation, including cAMP, vasodilator-stimulated phosphoprotein (VASP) dephosphorylation, phosphoinositide 3-kinase, and Rap1B.47-51 Pharmacologic approaches have shown a role for the P2Y12 receptor in dense granule secretion, fibrinogen-receptor activation, P-selectin expression, and thrombus formation, identifying it as a central mediator of the hemostatic response.52-56 Both P2Y12 and P2Y1 are indirectly involved in platelet P-selectin exposure and formation of platelet–leukocyte conjugates, which leads to leukocyte-TF exposure.57-59 Although not activated by ADP, platelets possess a third purinergic receptor (P2X1), which is a fast adenosine triphosphate (ATP)-gated calcium channel receptor mainly involved in platelet shape change. Figure 53–7 shows in detail the platelet purinergic receptors. The most recent advances in antiplatelet therapy related to the inhibition of the P2Y12 receptors are focused on the availability of faster acting and reversible inhibitors.60-62
Figure 53–7.
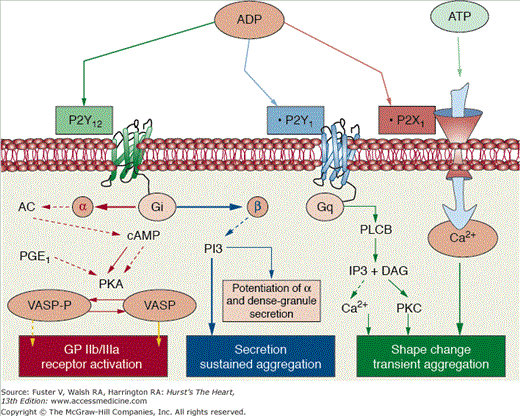
P2Y platelet receptors. P2Y receptors can be clearly divided into two subgroups: the Gq-coupled subtypes P2Y1 and the coupled to Gi P2Y12. P2Y1 that is responsible for platelet shape change and calcium mobilization is coupled to Gq and activates phospholipase Cβ (PLCβ) that is responsible for the formation of inositol (1,4,5)-trisphosphate (IP3) and diacylglycerol (DAG), an activator of protein kinase C (PKC). IP3 causes calcium mobilization from internal stores. The P2Y12 receptor couples primarily to Gατ2 inhibition of adenylyl cyclase (AC). The subsequent decrease in cAMP production leads, in turn, to a reduction in the activation of specific protein kinases (PKA), which can no longer phosphorylate the vasodilator-stimulated phosphoprotein (VASP); VASP phosphorylation is crucial for glycoprotein (GP) IIb/IIIa receptor inhibition. The subunit βγ activates phosphatidylinositol 3-kinase, which is an important signaling molecule for P2Y12-mediated platelet-dense granule secretion and GP IIb/IIIa receptor activation. Finally, P2X1 is a gated cation channel protein activated by adenosine triphosphate (ATP). This activation leads to increase intraplatelet calcium, platelet shape change, and a transient and weak platelet aggregation response. ADP, adenosine diphosphate. Reprinted from Ibanez et al.25 Under permission of the copyright holder: Oxford University Press.
During plaque rupture, in addition to platelet deposition in the injured area, the clotting mechanism is activated by the exposure of the plaque contents. The activation of coagulation leads to the generation of thrombin, which is a powerful platelet agonist in addition to being an enzyme that catalyzes the formation and polymerization of fibrin. Fibrin is essential in the stabilization of the platelet thrombus and its ability to withstand removal forces by flow, shear, and high intravascular pressure. The efficacy of fibrinolytic agents demonstrates the importance of fibrin in thrombosis associated with myocardial infarction (MI).
The blood coagulation system involves a sequence of reactions integrating zymogens (proteins susceptible to activation into enzymes via limited proteolysis) and cofactors (nonproteolytic enzyme activators) into three groups: (1) the contact activation (generation of factor XIa via the Hageman factor), (2) the conversion of factor X to factor Xa in a complex reaction requiring the participation of factors IX and VIII, and (3) the conversion of prothrombin to thrombin and fibrin formation63 (Fig. 53–8A).

It has been suggested that glycosaminoglycans and sulfatides are the triggering surfaces for in vivo initiation of contact activation; however, the physiologic role of coagulation contact activation is unclear because the absence of Hageman factor, prekallikrein, or high-molecular-weight kininogen does not induce a clinically apparent pathology. On the contrary, factor XI deficiency is associated with abnormal bleeding. Activated factor XI induces the activation of factor IX in the presence of Ca2+. Factor IXa forms a catalytic complex with factor VIII on a membrane surface and efficiently activates factor X in the presence of Ca2+. Factor IX is a vitamin K–dependent enzyme, as are factors II, VII, and X; prothrombin; and proteins C and S.
Citrate is calcium quelant frequently used in studies of platelet–vessel wall interaction. Its anticoagulant properties are based on its action over calcium. It blocks not only the coagulation cascade because the coagulation reactions do not proceed further than the activation of factor XI because of their dependence on Ca2+ but also inhibits platelet activation because of the central role of calcium in platelet activation (see Fig. 53–4). Platelets may provide the membrane requirements for the activation of factor X, although the participation of cells of the vessel wall (in exposed injured vessels) has not been excluded. As such, endothelial cells in culture have been shown to support the activation of factor X.41 Factor VIII forms a noncovalent complex with vWF in plasma, and its function in coagulation is the acceleration of the effects of IXa on the activation of X to Xa. Absence of factor VIII or IX produces the hemophilic syndromes (Fig. 53–8B).
The TF pathway, previously known as the extrinsic coagulation pathway through the TF–factor VIIa complex in the presence of Ca2+ induces the formation of Xa. A second TF-dependent reaction catalyzes the transformation of IX into IXa. TF is an integral membrane protein that serves to initiate the activation of factors IX and X and to localize the reaction to cells on which TF is expressed. Other cofactors include factor VIIIa, which binds to platelets and forms the binding site for IXa, thereby forming the machinery for the activation of X, and factor Va, which binds to platelets and provides a binding site for Xa. The human genes for these cofactors have been cloned and sequenced. In physiologic conditions, no cells in contact with blood contain active TF, although cells such as monocytes and polymorphonuclear leukocytes can be induced to synthesize and express TF.
Activated Xa converts prothrombin into thrombin. The complex that catalyzes the formation of thrombin consists of factors Xa and Va in a 1:1 complex. This activation results in the cleavage of fragment 1.2 and formation of thrombin from fragment 2. The interaction of the four components of the “prothrombinase complex” (Xa, Va, phospholipid, and Ca2+) enhances the efficiency of the reaction.
Activated platelets provide a procoagulant surface for the assembly and expression of both intrinsic Xase and prothrombinase enzymatic complexes.64,65 These complexes respectively catalyze the activation of factor X to factor Xa and prothrombin to thrombin. The expression of activity is associated with the binding of both of the proteases factor IXa and factor Xa and the cofactors VIIIa and Va to procoagulant surfaces. The binding of IXa and Xa is promoted by VIIIa and Va, respectively, such that Va and likely VIIIa provide the equivalent of receptors for the proteolytic enzymes.66,67 The surface of the platelet expresses the procoagulant phospholipids that bind coagulation factors and contribute to the procoagulant activity of the cell.67
Thrombin acts on multiple substrates, including fibrinogen, factor XIII, factors V and VIII, and protein C in addition to its effects on platelets. It plays a central role in hemostasis and thrombosis. The catalytic transformation of fibrinogen into fibrin is essential in the formation of the hemostatic plug and in the formation of arterial thrombi. Thrombin binds to the fibrinogen central domain and cleaves fibrinopeptides A and B, resulting in the formation of fibrin monomer and polymer formation. The fibrin mesh holds the platelets together and contributes to the attachment of the thrombus to the vessel wall.
The control of the coagulation reactions occurs by diverse mechanisms, such as hemodilution and flow effects, proteolytic feedback by thrombin, inhibition by plasma proteins (eg, antithrombin III [ATIII]) and endothelial cell–localized activation of an inhibitory enzyme (protein C), and fibrinolysis (Fig. 53–9). Although ATIII readily inactivates thrombin in solution, its catalytic site is inaccessible while bound to fibrin; it may still cleave fibrinopeptides even in the presence of heparin. Thrombin has a specific receptor in endothelial cell surfaces, thrombomodulin, which triggers a physiologic anticoagulation system.68 The thrombin–thrombomodulin complex serves as a receptor for the vitamin K–dependent protein C, which is activated and released from the endothelial cell surface. Thrombin generated at the site of injury binds to thrombomodulin, an endothelial surface-membrane protein, initiating activation of protein C, which in turn (in the presence of protein S) inactivates factors Va and VIIIa and limits thrombin effects. Loss of Va decreases the role of thrombin formation to negligible levels. Thrombin stimulates successive release of both tPA and plasminogen-activator inhibitor type 1 (PAI-1) from endothelial cells, thus initiating endogenous lysis through plasmin generation from plasminogen by tPA with subsequent modulation through PAI-1. Thrombin therefore plays a pivotal role in maintaining the complex balance of initial prothrombotic reparative events and subsequent endogenous anticoagulant and fibrinolytic pathways. The pivotal role of thrombin in the different mechanisms exposed here is represented in Fig. 53–10.




