Coronary Blood Flow and Myocardial Ischemia: Introduction
Regulation of Coronary Blood Flow
The defining characteristic of the coronary circulation is the direct relationship existing between coronary blood flow and myocardial oxygen consumption. The physiologic rationale is based on the requirement for coronary blood flow to meet the energy requirements of the heart. The main parameters dictating cardiac oxygen consumption are heart rate (chronotropy), cardiac contractility (inotropy), and left ventricular (LV) wall stress. Whereas coronary perfusion at rest in humans is approximately 200 mL/min, it can increase up to 1000 mL/min on maximal exercise. The difference between values at rest and maximal levels of coronary blood flow represents the coronary blood flow reserve. The mechanisms by which the coronary bed adapts blood flow to the cardiac workload represent one component of coronary autoregulation, that is, the recruitment of the coronary blood flow reserve to match coronary blood flow (O2 supply) to energy needs (O2 demand). This is accomplished via metabolic byproducts and adenosine, but it can also be modulated through an integrated regulation of substance release from the endothelium or from the myocardium itself, neural control, myocardial compressive forces, and aortic perfusion pressure. First, the myogenic tone of the small-caliber coronary resistance vessels is controlled by endothelial factors (such as nitric oxide), metabolic products (such as adenosine), and the autonomic nervous system (both cholinergic and α- and β-adrenergic receptors). Second, this chemical control of coronary blood flow is additional to the control exerted by physical forces (aortic perfusion pressure and intramyocardial compression forces). All of these components will be discussed in the first part of this chapter.
In contrast to most other vascular beds, the myocardium is perfused mainly during diastole and shows a sharp decrease in perfusion during systole, which can be attributed to myocardial compression (Fig. 54–1). The myocardial compressive forces are largely responsible for this phasic nature of coronary perfusion throughout the cardiac cycle. Because of this, a measurement of mean coronary vascular resistance is less meaningful than a calculation of vascular resistance at end-diastole prior to atrial contraction when compressive forces are minimal. A fall of coronary perfusion at the onset of ventricular systole is due to the squeezing forces of the contracting myocardium, when the intraventricular pressure that opposes coronary blood flow is roughly equal to the aortic perfusion pressure. Blood flow through large coronary arteries can even be transiently reversed during early systole. Reciprocally, ventricular relaxation during diastole is accompanied by a decrease in intraventricular pressure that is far greater than the decrease in aortic pressure, which allows optimal coronary perfusion early in diastole. In addition, the position of the coronary ostia just above the aortic valve leaflets favors the perfusion of the coronary bed during diastole by retrograde blood flow from the ascending aorta. As a consequence, systole contributes <20% of the total coronary blood flow during a cardiac cycle at rest. However, the fraction of coronary blood flow during systole can increase, particularly when tachycardia compromises diastole, as during electrical pacing or exercise (Fig. 54–2).
Figure 54–1.
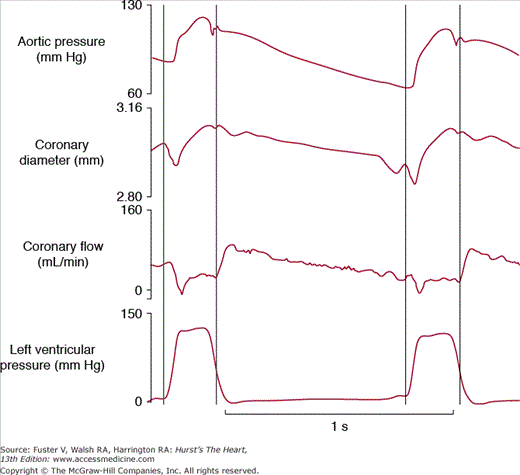
Phasic nature of coronary blood flow during the cardiac cycle. This figure illustrates the profile of aortic pressure, coronary diameter, coronary blood flow, and left ventricular pressure during the cardiac cycle in the conscious dog. Vertical lines indicate the duration of systole, and indicate that the large coronary arteries follow arterial pressure and are at a maximum, whereas the coronary blood flow is minimal during systole due to extravascular compression. Reproduced with permission from Vatner SF, Pagani M, Manders WT, Pasipoularides AD. Alpha adrenergic vasoconstriction and nitroglycerin vasodilation of large coronary arteries in the conscious dog. J Clin Invest. 1980;65:5-14.
Figure 54–2.
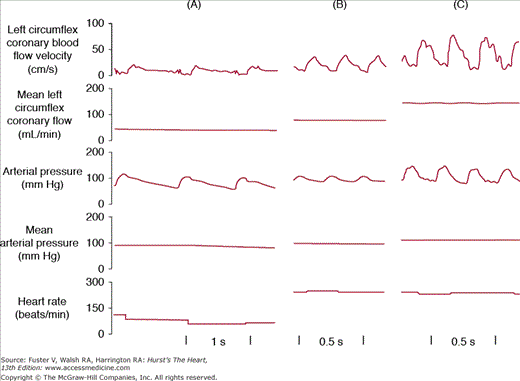
Regulation of coronary blood flow and heart rate by exercise. Comparison of coronary blood flow, aortic pressure, and heart rate in a dog at rest, during an atrial stimulation at a frequency of 248 beats/min, and during exercise at the same atrial frequency. Note that roughly one-third of the coronary hyperemia of exercise can be ascribed simply to the increase in heart rate. Reproduced with permission from Vatner SF, Higgins CB, Franklin D, Braunwald E. Role of tachycardia in mediating the coronary hemodynamic response to severe exercise. J App Physiol. 1972;32:380-385.
In a normal heart, the blood flow to the subendocardium is approximately 125% of that in the subepicardium,1 that is, the subendocardial–to-superepicardial (endo-epi) ratio, one index of myocardial ischemia, is above unity in the normally perfused heart. However, this ratio decreases dramatically in conditions of reduced perfusion pressure or during epicardial coronary artery constriction or occlusion, particularly in situations with well-developed coronary collaterals, which favor the subepicardium. In that condition, the maintained subepicardial perfusion is at the expense of the subendocardium and represents the basis of subendocardial ischemia. A reversed endo-epi flow ratio is the hallmark of myocardial ischemia. Furthermore, a vasodilator (eg, isoproterenol) can increase blood flow to the subepicardium at the expense of the subendocardium, a condition referred to as coronary steal. Another condition in which the subendocardial coronary blood flow will decrease, but not to the same extent as in ischemia, is cardiac hypertrophy. In this setting, not only do the coronary arteries use their mechanism of autoregulation due to the higher energy demand of the hypertrophied heart, but also the compressive forces become much larger, especially in the subendocardium.2
As mentioned earlier, the compressive forces are largely responsible for the phasic nature of coronary blood flow. The subendocardium is the region most vulnerable to changes in physical forces. Not only is the subendocardium submitted to the highest compressive force during systole, but also the subendocardial flow can be impaired during diastole in conditions of increased LV wall stress, such as during the advanced stage of cardiac hypertrophy (eg, as might accompany longstanding hypertension) or during heart failure. An important distinction must be made for coronary blood flow to the left ventricle and right ventricle due to compressive forces, which are lower in the right ventricle. Correspondingly, the fraction of systolic coronary blood flow is slightly higher to the right ventricle than to the left ventricle, whereas the total flow is less in the right ventricle due to the lower level of wall stress.3
Coronary oxygen extraction is almost maximal at rest. Myocardial oxygen extraction at rest averages 75% to 80%, and thus, it may increase only by a slight margin, up to a maximum of 90%, upon maximal demand, such as heavy exercise. As a consequence, the main mechanism to bring more oxygen to the myocardium upon increased demand is to increase coronary blood flow.
The primary determinant of coronary blood flow is myocardial oxygen consumption (MVO2),4 which is calculated as the product of coronary blood flow and the arteriovenous (A-V) O2 difference. As noted earlier, because A-V O2 difference cannot increase greatly, an increase in MVO2 must be accompanied by an increase in coronary blood flow. Because cardiac work is tightly coupled to MVO2, the fact that heart rate increases three-fold and contractility increases five-fold, along with increases in LV wall motion during maximal exercise, results in a requirement for a five-fold increase in coronary blood flow observed in that condition. Thus, the main determinants of MVO2 are the factors primarily involved in regulating myocardial metabolic demand (heart rate and contractility) and LV wall stress.
Heart rate is the principal mechanism by which cardiac output can be increased. In humans, heart rate increases by approximately three-fold during intense exercise due to an adrenergic stimulation from both neural and circulating catecholamines.5 Heart rate is probably the factor that affects MVO2 the most. Increasing the heart rate to the same level as reached during exercise in an experimental preparation will increase coronary blood flow by 50%.6 During intense exercise, heart rate accounts for approximately 30% of the increase in coronary blood flow (see Fig. 54-2). In the case of tachycardia, the oxygen demand increases not only strictly because of a higher heart rate, but also because tachycardia decreases the diastolic time of coronary perfusion. Clinically, the Framingham study has shown that individuals with higher heart rates at rest have a higher risk of cardiovascular mortality.
The increase in contractile state of the myocardium is critical for adjustments to exercise and response to sympathetic stimulation. For example, during intense exercise, LV dP/dt (rate of change of LV systolic pressure), an index of contractility, can also increase four- to fivefold, which permits stroke volume to increase despite increases in heart rate. This increase in myocardial contractility also increases MVO2, which requires a commensurate increase in coronary blood flow.
LV systolic wall stress correlates directly with MVO2. The formula for LV wall stress demonstrates that this critical determinant of MVO2 is directly proportional to LV pressure and LV diameter or volume and inversely proportional to LV wall thickness. Accordingly, with LV hypertrophy due to pressure overload, which increases LV systolic pressure, the wall stress is normalized by the LV hypertrophy (ie, increased wall thickness). For that reason, baseline myocardial blood flow per gram of tissue remains essentially at normal levels (1.0 mL/min/g) in LV hypertrophy. For the same level of ventricular hypertrophy (ie, where wall thickness is reduced by LV dilation), MVO2 will increase much more than in a condition of normalized wall stress.7 In patients with systemic hypertension, for example, MVO2 increases, compared with normotensive subjects, before wall stress is normalized. This increased oxygen requirement implies a recruitment of the coronary blood flow reserve already at rest.
As developed by Berne and colleagues, the most potent stimulus that suppresses the myogenic tone and thus dilates the coronary vasculature is adenosine.8 The ratio between the coronary blood flow after administration of adenosine and the flow at rest represents the coronary blood flow reserve. In normal individuals, this ratio is between 4 and 5, with the flow ranging from 200 mL/min at rest to 1000 mL/min at maximal exercise in well-trained athletes. In cardiac myocytes in vivo, adenosine is formed when the rate of ATP degradation exceeds the rate of ATP regeneration. This biochemical mechanism is extremely sensitive because the concentration of adenosine in cardiac tissue is approximately 1000-fold lower than the concentration of adenosine triphosphate (ATP). Therefore, even a minute imbalance between ATP degradation and synthesis will result in an increase of adenosine that will be qualitatively, if not quantitatively, very significant. Adenosine production is triggered by hypoxia, either in conditions of ischemia or when oxygen demand increases, such as during exercise. The adenosine hypothesis stipulates that the adenosine released from the myocytes binds to A2 receptors on the smooth muscle cells of the coronary bed, which stimulates vasodilation through production of cyclic adenosine monophosphate.9 The resulting decrease in vascular resistance increases the coronary blood flow, which restores the balance between oxygen demand and supply. The same adenosine that is formed under these conditions will bind A1 receptors on the cardiac myocytes to decrease inotropy and will trigger the mechanisms of preconditioning.10 Inhibition of adenosine receptors does not abolish the coronary autoregulation in absence of ischemia, suggesting that, in physiologic conditions, the endothelial control of coronary blood flow predominates, whereas, in ischemic conditions, the metabolic control takes over through the production of adenosine. However, nitric oxide production increases in conditions of ischemia11 and may therefore be complementary to the effects of adenosine.
Coronary autoregulation represents a mechanism by which the coronary artery can maintain a constant flow that matches the metabolic demand independently of the perfusion pressure. The coronary blood flow can increase up to fivefold between the basal state and the maximal perfusion, which represents the coronary blood flow reserve. Therefore, coronary autoregulation represents a mechanism by which the coronary blood flow reserve can be recruited to maintain the desired perfusion level. It is important to note that there are two distinct definitions of coronary autoregulation: the coronary autoregulation controlled by the perfusion pressure and the coronary autoregulation that is an adaptation of coronary resistance to the metabolic demand. These two components, or definitions, of coronary autoregulation are discussed next.
Coronary autoregulation is a mechanism intrinsic to myogenic tone in the coronary vessels by which the coronary artery maintains a constant blood flow in the face of variations in perfusion pressure.12 The vascular smooth muscle cells making the tunica media of the coronary arteriole are rich in stretch-operated ion channels activated by shear stress and that stimulate cell contraction. The high myogenic tone that results from the contractile activity of the smooth muscle cells translates into a high resistance to flow. This process allows the flow to be kept constant when the perfusion pressure varies between 40 and 140 mm Hg.12 These values were first established in experimental models and subsequently confirmed in humans. Consequently, in cases of severe coronary artery stenosis or shock, when the vascular resistance has fallen to its maximal capacity, the flow becomes directly proportional to the perfusion pressure.
The main cause of coronary stenosis and obstruction of a coronary artery is atherosclerosis. The single most important parameter of a coronary stenosis is the extent of reduction of the internal luminal diameter because, according to the laws of hydrodynamics, the resistance created by the stenosis is proportional to the fourth power of the minimal luminal diameter. In other words, even a slight decrease in diameter will result in a major loss of pressure. As an example, it has been calculated that an atherosclerotic plaque progressing from an 80% to a 90% obstruction of the vessel diameter would result in a three-fold increase in coronary resistance. The effect of coronary obstruction on blood flow therefore depends largely on the extent of the stenosis. For example, although minor obstructions do not have hemodynamic repercussions, a coronary stenosis obstructing at least 40% of the vessel diameter results in a decrease of perfusion pressure, which requires a matching decrease of coronary resistance to maintain the normal flow at rest.13 The decreased resistance is mediated mainly by adenosine release from the myocardium supplied by the obstructed artery. This process requires recruiting the coronary blood flow reserve already at rest and therefore limits the capacity to exercise. If the obstruction progresses, the coronary blood flow reserve will progressively decrease. When the coronary stenosis becomes so severe that it occludes at least 80% of the vessel diameter, the coronary reserve will be totally recruited at rest, and at that point, any further obstruction will result in a limitation of blood flow already at rest. This concept is further developed later in the Coronary Blood Flow Reserve section, which describes in more detail the coronary blood flow reserve.
The endothelium both produces vasoactive substances (eg, nitric oxide, prostacyclin, endothelin-1, epoxyeicosatrienoic acids) and responds to circulating factors (eg, epinephrine, serotonin). The overall function of the endothelium is to increase the flow through the arteries, if we exclude the tonic vasoconstriction induced by excessive production of endothelin-1. Therefore, endothelial dysfunction resulting from endothelial damage that accompanies most risk factors will result in increased vascular resistance and impaired flow. The most potent vasodilator released by the endothelium is nitric oxide (NO), which is produced by the stimulation of the cholinergic cascade and which immediately diffuses as a gas from the endothelium to the surrounding vascular smooth muscle cells where it induces a cyclic guanosine monophosphate (cGMP)–dependent muscular relaxation.14 Paradoxically, acetylcholine provokes vasoconstriction in isolated coronary arteries in vitro. These apparently contradictory results can be reconciled in light of the seminal observation by Furchgott and Zawadzki15 that acetylcholine induces vasoconstriction, rather than vasodilation, in isolated aortic rings denuded of endothelial cells, but in vessels with endothelium intact, acetylcholine induces prominent vasodilation. NO is the main mediator of the vasodilatory effect of acetylcholine (which is released physiologically through the parasympathetic neural stimulation), but its production is also increased by stimulation of the α-adrenergic receptors. In addition to NO, another potent vasodilator released by the endothelium is prostacyclin, which is involved in the normal vasodilation at rest and in the adaptation to increased shear stress. Blocking prostacyclin synthesis with cyclooxygenase inhibitors (eg, aspirin, indomethacin, other nonsteroidal anti-inflammatory drugs) can reduce the coronary blood flow at rest in patients.
The release of NO by stimulation of endothelial cells by acetylcholine (the parasympathetic neurotransmitter) remains one of the most important mechanisms of vasodilation throughout the vasculature, which is reflected by the use of nitrates to decrease the preload in patients with coronary artery disease. Vagal stimulation has a vasodilatory effect in the coronary bed, thereby increasing coronary blood flow. In addition to a direct effect on the coronary bed, increased parasympathetic tone reduces heart rate, arterial pressure, and inotropy, which altogether reduce the oxygen demand of the myocardium.16 However, when these parameters are kept constant in experimental preparations where both heart rate and perfusion pressure can be controlled, vagal stimulation still increases coronary blood flow. This effect is reproduced by administration of acetylcholine and is blocked by atropine. It is now well established that endothelial dysfunction and reduced NO production, which often accompany atherosclerosis, increase the frequency of coronary vasospasm. Even if the coronary obstruction resulting from the atherosclerotic plaque is minor, the risk of vasospasm in that condition may be sufficient to induce ischemia and even myocardial infarction.
The stimulation of cardiac sympathetic nerves markedly increases coronary blood flow due to the increased metabolic demand induced by the increase in workload (heart rate, contractility, LV wall stress) resulting from catecholamine release. This increased metabolic demand is blocked by pretreatment with a β- adrenergic antagonist, which unmasks a vasoconstrictive effect.17 This vasoconstriction can in turn be blocked by an α-adrenergic antagonist.18 These results indicate that sympathetic stimulation in vivo increases heart rate and inotropy mainly through the β-adrenergic receptors, whereas stimulation of the α-receptors induces a vasoconstrictive response. Similarly, direct stimulation of α1-adrenergic receptors with pharmacologic agonists can provoke vasoconstriction in coronary vessels. An important consequence of the α-adrenergic–mediated vasoconstriction in a context of increased workload is an increase in oxygen extraction,19 which might be particularly relevant during exercise. The vasoconstriction resulting from α-adrenergic receptor stimulation is increased in patients with coronary artery disease,20 probably reflecting the partial use of the coronary blood flow reserve in that situation. The α-adrenergic tone of the coronary arteries can be activated by stimulation of reflex pathways, such as carotid baroreceptors upon carotid sinus hypotension, as well as by direct sympathomimetic stimulation. Similarly, stimulation of the carotid chemoreceptor, pulmonary inflation reflex, and arterial baroreceptor hypertension induce coronary vasodilation that results from a withdrawal of the α-adrenergic tone, potentially with a smaller vagal component contributing to the vasodilation (Fig. 54–3).
Figure 54–3.
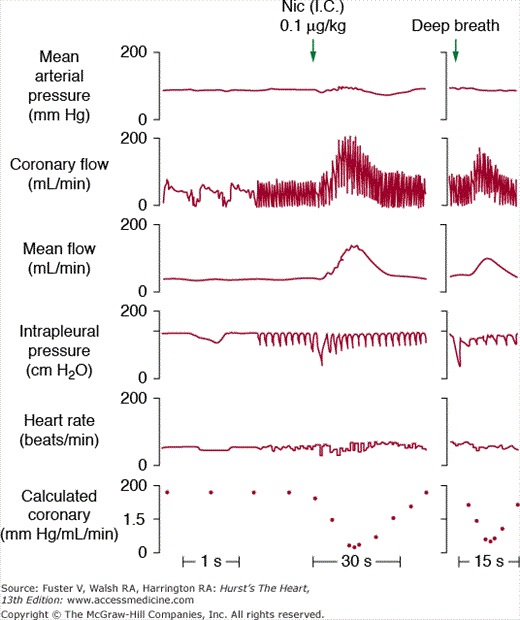
Reflex adaptation of coronary blood flow. Effects of intracarotid (I.C.) administration of nicotine (Nic) at a dose that has no systemic effect but that stimulates the carotid chemoreflex resulting in increases in respiration depth as reflected by the change in intrapleural pressure and marked increase in coronary blood flow and decreases in collateral coronary vascular resistance. The reflex coronary vasodilation can be mimicked by a spontaneous deep breath (right panel), which stimulates the pulmonary inflation reflex. The effect of nicotine can be reproduced by an episode of spontaneous deep breath. Reproduced with permission from Vatner SF, McRitchie RJ. Interaction of the chemoreflex and the pulmonary inflation reflex in the regulation of coronary circulation in conscious dogs. Circ Res. 1975;37:664-673.
Stimulation of the β-adrenergic receptors achieves coronary vasodilation through two complementary mechanisms. Binding of the receptors in the coronary vessel itself will stimulate a Gs protein–mediated relaxation of the vascular smooth muscle cells.21 In addition, binding of the receptors on the cardiac cells will stimulate a Gs protein–mediated increase in contractility, which, together with the increase in heart rate, will increase the metabolic demand and activate the recruitment of the coronary blood flow reserve through adenosine release. Whereas the β2-receptors are mainly responsible for the direct vasodilatory mechanism, the contribution of the β1-receptor predominates for the metabolic effect resulting from increased workload.22 As a consequence, treatment of patients with β-blockers will reduce coronary blood flow and may exacerbate coronary vasospasm. However, the negative inotropy and reduction of heart rate following administration of β-blockers markedly reduce the risk of exercise-induced angina and provide protection following myocardial infarction and, accordingly, are used widely in patients with ischemic heart disease and hypertension.
Under normal circumstances, large coronary arteries participate little in the regulation of coronary resistance because the chemical control of coronary blood flow is mainly performed at the level of small-caliber resistance vessels. However, the large coronary arteries play a major role in the regulation of coronary blood flow in pathologic settings, especially in conditions of coronary vasospasm and obstruction by an atherosclerotic plaque, when blood flow becomes largely dependent on perfusion pressure, and by shear stress (flow-dependent regulation of flow). The physical components of coronary blood flow include the coronary perfusion pressure, flow-dependent regulation, and control by neurohumoral mechanisms. In contrast to the phasic measurement of coronary blood flow, the phasic measurement of large coronary artery diameter parallels that of aortic pressure (see Fig. 54–1).
NO production is also increased by shear stress, and therefore, it represents an excellent mediator of autoregulation in conditions of increased cardiac output, such as during exercise.23 This mechanism represents the molecular basis of flow-dependent coronary vasodilation. The increased shear stress in conditions of increased flow, which is mainly determined by the viscosity of the blood and its velocity, represents a stimulus for NO production that is followed by vasodilation and subsequent decrease in shear stress. One of the first investigations was by Hintze and Vatner,24 who observed that following a temporary coronary artery occlusion during the reactive hyperemia phase, the large coronary arteries dilated (termed reactive dilation), which was entirely dependent on the increase in coronary blood flow.
The best experimental method to gauge the coronary blood flow reserve is by producing a transient occlusion of a large coronary artery. Upon reperfusion, the coronary blood flow reserve is fully recruited, inducing transiently a maximal flow called reactive hyperemia,25 which is accompanied by an increased diameter of the coronary artery (reactive dilation),24 discussed earlier. Whereas the reactive hyperemia results mainly from adenosine release, the reactive dilation is due to the NO-mediated response to increased shear stress following the increase in flow due to the hyperemia. This experiment shows that an obstacle to coronary blood flow is the most potent mechanism to challenge the system of autoregulation. When this system reaches its limits in the resistance vessels, the flow becomes mainly dependent on the perfusion pressure in the large coronary arteries.
Coronary perfusion pressure is directly proportional to the aortic pressure. It has been known for over a century that increasing the perfusion pressure in an isolated preparation has a positive inotropic effect on cardiac contractility, a phenomenon known as the Anrep effect.26 However, changes in aortic pressure are not necessarily accompanied by parallel changes in coronary blood flow because the coronary circulation adapts its resistance primarily to the oxygen needs. This is due to the fact that aortic perfusion pressure also represents the cardiac afterload, an important determinant of LV wall stress and MVO2. Any increase in aortic pressure will increase the metabolic demand of the myocardium. Therefore, variations in vascular resistance, rather than changes in perfusion pressure, are the main mediators of flow autoregulation. For example, coronary blood flow can increase up to five-fold in conditions of maximal exercise despite a limited increase in aortic pressure because of the combined vasodilatory effects of metabolic and endothelial effectors. Reciprocally, a decrease in aortic pressure provokes an increase in heart rate, which may also activate the mechanisms of metabolic autoregulation, as well as the myogenic component through the change in pressure and myogenic tone.
Large coronary arteries react to both cholinergic and adrenergic stimulation. Similarly to the resistance vessels, nitroglycerin induces a marked vasodilation of large arteries, whereas α-adrenergic stimulation results in vasoconstriction and β-adrenergic stimulation results in vasodilation. However, the large coronary arteries are relatively more sensitive to the α-adrenergic–mediated vasoconstriction than the resistance vessels. Interestingly, α-adrenergic stimulation induces vasoconstriction despite an increase in both preload and afterload that should theoretically result in a pressure-induced dilation of the coronary artery. The β-adrenergic vasodilation of the large arteries is also specific (ie, mediated by vascular receptors) because it can be dissociated from changes in other cardiac parameters, such as heart rate.21 The cholinergic effect is reversed in conditions of endothelial dysfunction, according to Furchgott’s paradigm.15
The coronary blood flow reserve represents the difference between coronary blood flow at rest and maximal perfusion. The main condition recruiting the coronary blood flow reserve in the normal heart is exercise. However, this reserve is already recruited at rest in the diseased heart, in conditions such as ischemic heart disease.
The massive increase in myocardial metabolic demands during exercise requires a matching increase in coronary blood flow because, as mentioned earlier, coronary oxygen extraction is almost maximal already at rest, and the maximal increases in heart rate, LV wall stress, and myocardial contractility during exercise increase MVO2 and secondarily coronary blood flow. As a result, the only mechanism to bring more oxygen to the myocardium upon increased demand is an increase in flow (see Fig. 54–2). All of the mechanisms of coronary blood flow regulation will participate in this adaptation. The increased oxygen demand results from an increase in heart rate, cardiac contractility, and ventricular wall stress from the adrenergic stimulation. Variations of pressure in large coronary arteries during exercise are intertwined with variations of resistance in smaller vessels. For instance, an increase in heart rate will shorten the time of ventricular relaxation during which the perfusion pressure is optimal, but a metabolic effect usually accompanies this situation to maintain or increase the coronary blood flow.27 During exercise, the contribution of systole to the coronary blood flow increases to approximately 40%, whereas it represents only 15% to 20% at rest. During exercise, coronary resistance decreases through the three mechanisms described earlier (ie, flow-mediated dilation and NO release, metabolic dilation by adenosine, and β-adrenergic vasodilation). It has been demonstrated experimentally that exercise is also accompanied by a vasoconstrictive α-adrenergic stimulation.18 Although counterintuitive, this mechanism could balance an excessive decrease in coronary resistance and could also improve myocardial oxygen extraction. In addition, the aortic perfusion pressure also increases, thereby improving the flow through large coronary arteries.
The considerations detailed in the previous section apply only to the normal heart. For example, limited coronary reserve in the hypertrophied heart severely impairs its capacity to exercise. In a canine model of both right ventricular and LV hypertrophy, it was demonstrated that the animals present a massive postexercise coronary hyperemia, suggesting a perfusion deficit of the hypertrophied ventricle during exercise.
Ischemic heart disease is another pathologic condition that limits the coronary blood flow reserve. The purpose of the coronary blood flow reserve in ischemic heart disease is to maintain a normal flow at rest for as long as possible. As mentioned earlier, a coronary obstruction that occludes at least 40% of the vessel diameter will recruit the coronary blood flow reserve to maintain a normal flow in rest conditions. Above that threshold of 40%, the coronary blood flow reserve is recruited to an extent that is proportional to the severity of the stenosis. When patients with coronary obstruction start exercising, the increased energy demand will not be matched properly by an increased oxygen supply because the coronary blood flow cannot increase further proportionately to the metabolic needs, which may trigger an episode of exercise- or stress-induced angina. If the atherosclerotic plaque obstructs at least 80% of the vessel diameter (which corresponds to approximately 95% of the cross-sectional area), the coronary blood flow reserve will be compromised at rest. Coronary blood flow will be directly related to perfusion pressure.28 If the obstruction becomes any worse than 80% (eg, when a thrombus forms on an ulcerated plaque), the coronary blood flow will become insufficient even at rest, which results in episodes of unstable angina (pain in absence of increased workload). This situation is “unstable” because a thrombotic plaque can rapidly evolve into total coronary occlusion and subsequent myocardial infarction.
Besides the mechanical obstacle created by the plaque, other factors will limit the blood flow in an atherosclerotic artery. An additional reason for the lack of increased flow in response to increased shear stress in the diseased artery is the fact that endothelial dysfunction frequently is superimposed on atherosclerosis, which limits the recruitment of NO-mediated vasodilation. In all cases, the coronary blood flow reserve in the subendocardium will be recruited faster than in the subepicardium. There will be a point at which the subendocardial arterioles are fully dilated (and therefore, the subendocardial flow becomes directly proportional to the perfusion pressure), whereas the subepicardial arteries retain some autoregulation.29 At that point, any further dilation in the subepicardium will redistribute the flow from the subendocardium to the subepicardium (coronary steal) and exacerbate subendocardial ischemia. In addition, in case of thrombus formation, the serotonin released by activated platelets will also promote local vasospasm in a context of endothelial dysfunction. Thus, limitations of coronary blood flow reserve become an integral component of coronary artery disease and of the mechanisms of myocardial ischemia that are described in the following section.
Mechanisms of Reversible Myocardial Ischemia
Myocardial ischemia is the result of an imbalance between myocardial oxygen supply and myocardial oxygen demand and has a variety of consequences (Fig. 54–4) depending on the severity of the reduction in coronary blood flow, the length of the ischemic insult, the area of myocardium subserved by the occluded artery, and the presence of collateral vessels from other adjacent vascular beds within the heart. These consequences of ischemia include angina pectoris, myocardial stunning, myocardial hibernation, pre- or postconditioning, or under the most severe instances, a myocardial infarction with subsequent heart failure. Of course, the hemodynamic determinants of oxygen demand will also influence the outcome and include heart rate, wall stress, and myocardial contractility. Although a reduction or abolition in coronary blood flow is thought to usually be detrimental to the myocardium, there are instances where brief periods of coronary artery occlusion followed by intermittent periods of reperfusion prior to a more prolonged coronary occlusion are beneficial to the heart and make it resistant to stunning or infarction, a phenomenon termed myocardial or ischemic preconditioning (IPC).30 There is also evidence that suggests that an IPC protocol administered immediately upon reperfusion can also result in a reduction in infarct size as compared with a control group, a phenomenon termed postconditioning (POC).31 Both of these potentially clinically applicable phenomena will be discussed in more detail later in this chapter.
Figure 54–4.
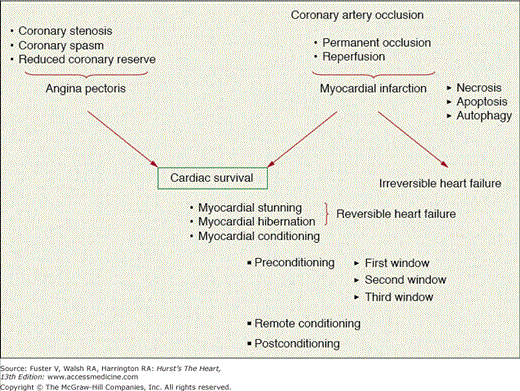
Paradigms of myocardial ischemia. Coronary stenosis leads to the different forms of angina and is reversible upon reperfusion by angioplasty or coronary bypass surgery. Repetitive bouts of ischemia upon coronary stenosis can lead to the formation of survival syndromes (stunning, hibernation, and conditioning). Coronary occlusion also leads to myocardial infarction, which will evolve into heart failure in absence of early reperfusion, or leads to survival mechanisms if reperfused.
Myocardial ischemia does not generally occur via a total coronary artery occlusion but usually results from one of two sequelae of events, which result in the clinical syndrome of angina pectoris. In the majority of cases, a single stenosis or multiple flow-limiting coronary artery stenoses are present in one or more of the major epicardial coronary arteries, and no symptoms of ischemia occur at rest but often occur when the patient exercises or is under other potentially stressful conditions (ie, oxygen demand exceeds oxygen supply). This situation is often termed secondary angina because the ischemia is secondary to an increase in oxygen requirements and is relieved when the patient stops exercising, removes himself or herself from the stressful situation, or a rapidly acting vasodilating drug is administered, such as sublingual nitroglycerin. These patients usually present with ST-segment depression on the electrocardiogram suggesting subendocardial ischemia. Subendocardial ischemia normally occurs in secondary angina because of the worsening of a transstenotic pressure gradient, which does not generally produce ischemia at rest because normal flow is maintained by a compensatory dilation distal to the stenosis. As described in the first part of the chapter, it generally requires a reduction in the resting epicardial diameter of 85% to 90% of a major coronary artery to produce regional ischemia at rest. Usually, the acute increase in blood flow that occurs across a flow-limiting stenosis during stress or exercise will markedly reduce the poststenotic pressure, which is the major determinant of blood flow to the subendocardium. Along with the increase in the transstenotic pressure gradient and reduction in subendocardial perfusion, an acute increase in LV filling or end-diastolic pressure may also occur because of regional ischemia, and this may result in a further reduction in the poststenotic diastolic perfusion pressure and subendocardial perfusion, a condition that has been termed diastolic crunch. In the presence of a prolonged flow-limiting stenosis and intermittent episodes of ischemia, coronary artery collaterals from other vascular beds may be stimulated to grow and the distal coronary pressure may increase, thus increasing the coronary blood flow reserve to the affected area and reducing the significance of the flow-limiting stenosis and increasing the anginal or ischemic threshold.
Another mechanism that has been shown to be responsible for ischemia distal to a flow-limiting coronary artery stenosis is the coronary steal described earlier in this chapter. This situation is present when vasodilation occurs in a coronary bed with a much greater coronary blood flow reserve than an adjacent coronary bed. This may occur during exercise or in the presence of certain arteriolar vasodilators and may result in steal from the subendocardium to the subepicardium, which is termed transmural steal, or from areas perfused by collaterals where dilation of an artery feeding the collaterals may have a stenosis, and poststenotic dilation will decrease the collateral perfusion pressure and collateral flow. This scenario is termed lateral coronary steal. Generally, steal does not occur in the presence of currently used antianginal drugs such as the nitrates or calcium channel blockers because these drugs have been shown to selectively dilate large coronary conductance vessels as opposed to the more distal arterioles.
Collateral blood vessels are stimulated to grow as a result of the poststenotic pressure gradient, which develops as a result of atherosclerosis in the large epicardial coronary arteries, and/or as a result of the intermittent episodes of ischemia, which develop as a result of single or multiple flow-limiting coronary artery stenoses present in the vascular bed. Generally, these collaterals develop from small preexisting anastomoses that are preformed and connect the vascular beds, although in some cases, these vessels appear to be newly formed. The collateral circulation is very species dependent, with numerous collaterals being found in guinea pig hearts, less in dogs and humans, and essentially none in other animal species such as rabbits, pigs, rats, sheep, mice, and nonhuman primates. In young humans, before coronary artery disease has progressed, collaterals are also sparse. However, in older patients, coronary artery disease has largely induced collateral formation. These preexisting anastomoses progressively form over several months to years by increasing in width and developing a smooth muscle coat and endothelial cells and may reach diameters of up to 200 to 400 μm. These vessels may proliferate to the point where they may prevent ischemia in vascular beds distal to a total coronary artery occlusion at rest or during mild stress.
The second major type of angina is the result of spasm of a major coronary artery and has been termed primary or variant angina because the ischemia occurs as the result of a primary decrease in oxygen supply (ie, coronary blood flow) without a change in myocardial oxygen demand. This syndrome was originally noted by Myron Prinzmetal and colleagues and is often referred to as prinzmetal angina (see Chap. 59). In some instances, there has been evidence that some patients may have microvascular spasm or thrombosis that is responsible for the ischemia and symptoms, but these are more difficult to diagnose by commonly used angiographic techniques. Coronary spasm can occur in an area of the artery with or without the presence of a stenotic lesion detectable by angiography.32,33 Coronary spasm may lead to ST-segment elevation on the electrocardiogram, which is indicative of severe transmural myocardial ischemia and threatened infarction. In some cases, subendocardial ischemia and ST-segment depression are observed if the occlusion is not complete. Coronary spasm can be reproduced by the intracoronary administration of ergonovine into the affected segment of the vascular tree, which is relieved by intracoronary nitroglycerin. Similar to secondary angina, this syndrome can be quickly relieved by the sublingual administration of nitroglycerin or isosorbide dinitrate, which are effective vasodilators for large coronary arteries. Although the precise mechanism responsible for coronary spasm has not been identified, a number of vasoconstrictor substances have been proposed to be responsible, including sympathomimetic amines such as norepinephrine or epinephrine, serotonin, histamine, endothelin, thromboxane A2, and acetylcholine, which all act on G protein–coupled receptors. Increases in extracellular K+ or Ca2+, which would be expected to result in membrane depolarization and vasoconstriction, have also been implicated as causing spasm. That Ca2+ channel blockers are the mainstays in therapy in preventing coronary spasm supports the possibility that these latter two mechanisms are key components of spasm.
Unstable angina is characterized by a sudden worsening of chronic angina in which angina may appear at rest or with exertion but with an increased frequency and severity. These patients are often prone to develop an acute myocardial infarction as a result of plaque rupture, thrombosis, and coronary vasoconstriction or spasm. Another feature of unstable angina is a reduction in the ischemic threshold during exercise, which is suggestive of an increased severity of a flow-limiting stenosis. A full review of the clinical issues associated with the acute coronary syndromes can be found in Chap. 56.
Some patients with angiographically normal coronary arteries have a positive exercise stress test with ST-segment depression on the electrocardiogram and perfusion defects indicative of myocardial ischemia and classical angina pectoris. This condition is often termed cardiac syndrome X and most often occurs in postmenopausal women. Although the exact etiology of this syndrome is not certain, there is evidence that this may be the result of microvascular dysfunction particularly of endothelial cells. Evidence for microvascular dysfunction has been suggested by exercise-induced perfusion abnormalities by radionuclide imaging, a reduced LV ejection fraction during exercise compared with control patients, the presence of ST-segment changes indicative of subendocardial ischemia during exercise, and the perception of classical anginal pain during exercise, which may persist for up to 30 minutes after exercise. These patients often respond poorly to sublingual nitrates and demonstrate a variable response to β-receptor blockers and calcium antagonists. In one study, women with syndrome X had impaired vasodilator responses to acetylcholine, an endothelium-dependent vasodilator, and to sodium nitroprusside, an endothelium-independent vasodilator, which clearly suggested a generalized impaired vasodilator function in these patients.34 However, other investigators have failed to show microvascular dysfunction uniformly in patients with syndrome X and have suggested that other possibilities exist to explain the pain that these patients feel, including an abnormality in pain perception, estrogen-related hormonal irregularities, insulin resistance, and a psychological basis. Thus, although microvascular dysfunction appears to be involved in many of these patients, this syndrome may be multifactorial, and a comprehensive treatment regimen may be necessary in treating this disorder.
There are a number of other consequences that occur when the myocardium is subjected to brief periods of myocardial ischemia and reperfusion or to prolonged low flow. These syndromes have been termed myocardial stunning and hibernating myocardium, respectively. The consequences and potential mechanisms of these two syndromes on myocardial cell injury will be described later in this chapter.
The primary manifestation of myocardial ischemia is angina pectoris, irrespective of the type of angina and the causal factors involved. Unfortunately, myocardial ischemia also appears to occur in cases without the symptoms of chest pain, a syndrome known as silent ischemia, and anginal pain may occur in the absence of myocardial ischemia, possibly as a result of esophageal spasm or some other form of gastrointestinal disorder. Typically, the pain of angina occurs retrosternally and may manifest as a crushing, burning, or squeezing type of discomfort. The pain may spread to the lower part of the neck and radiate down the left arm or either arm in some cases. The pain may be mild or intense and persist for approximately 5 to 10 minutes and is usually relieved rapidly by the administration of nitroglycerin sublingually. If three sequential tablets of nitroglycerin do not relieve the pain, the pain is likely the result of an ongoing myocardial infarction or is from an extracardiac source. Chemical mediators such as adenosine, which is known to be released in large quantities during ischemia, are most likely responsible for the pain of angina or a myocardial infarction via activation of sensory nerves surrounding the coronary arteries. These nerves project to the dorsal roots of the spinal cord, where they converge on thalamic centers, and finally to the cortex. Unfortunately, nerves from other visceral organs have a similar distribution, and this may be responsible for nonspecific cardiac pain that is mistaken for angina in many cases.
Until the mid-1960s, myocardial ischemia was thought to result in irreversible myocyte damage. However, this concept was challenged with the discovery that early reperfusion may lead to contractile recovery of the ischemic territory. This proof of principle that ischemia does not necessarily mean cell death led to the unraveling of physiologic conditions in which ischemia-reperfusion boosts the survival capacity of the myocardium and fundamentally changed the clinical approach to the patient presenting with symptoms suggestive of acute myocardial ischemia (see Chaps. 56 to 61). The four most common conditions of cardiac cell survival in a context of ischemia-reperfusion are described as myocardial stunning, IPC, POC, and hibernating myocardium.
Heyndrickx and coworkers35 were the first to demonstrate that reversibly injured myocardium following a 5- or 15-minute episode of ischemia and reperfusion resulted in a deficit in regional contractile function in dogs following reperfusion that lasted for 6 hours following 5 minutes of ischemia and greater than 24 hours following 15 minutes of ischemia (Fig. 54–5). This phenomenon was termed myocardial stunning, which can be defined as a prolonged and fully reversible dysfunction of the ischemic heart that persists after reperfusion despite the normalization of blood flow. Another characteristic of myocardial stunning is that the affected area is responsive to inotropic stimulation by catecholamines such as dobutamine or noncatecholamine-positive inotropes such as milrinone without any deleterious consequences following inotropic support. In other words, the time course of stunning is not altered by the increase in contractile function produced by the positive inotropes.
Figure 54–5.
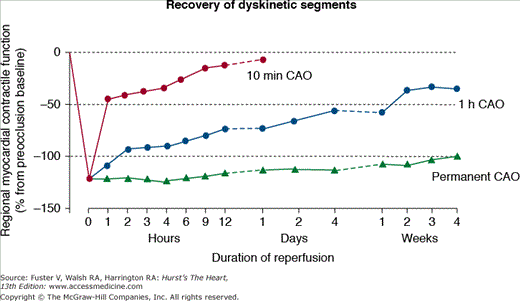
Importance of early reperfusion on postischemic function recovery. This figure compares the extent of recovery of cardiac contraction after a coronary artery occlusion (CAO) of 10 minutes or 1 hour, with reperfusion, compared with a permanent occlusion. The delayed recovery of function that occurs with reperfusion is due to myocardial stunning. Adapted from Depre C, Vatner SF. Mechanisms of reversible ischemic dysfunction. In: Walsh RA, ed. Molecular Mechanisms of Cardiac Hypertrophy and Failure. London, United Kingdom: Taylor & Francis; 2005:221-246.
Initially described in animal models, myocardial stunning also occurs in the clinical setting. Stunning can occur in a number of conditions including following exercise in the presence of a flow-limiting stenosis, following a brief period of total coronary occlusion such as might occur in patients with primary angina due to spasm, or following global ischemia after cardiopulmonary bypass. Stunning may even occur in combination with a myocardial infarction, where the subendocardium is infarcted and the overlying reversibly injured subepicardium may be stunned for a prolonged period of time, perhaps days to weeks. Characteristically stunned myocardium does not result in electrocardiographic abnormalities, and coronary blood flow to the stunned area is usually normal, which suggests that there is a flow-function mismatch. Myocardial stunning can occur due to the ischemic bout that is induced by percutaneous coronary intervention. The occlusion due to the balloon inflation, followed by reperfusion when the balloon deflates, triggers an ischemic attack, which, in turn, stuns the myocardium. However, the ischemic attack induced by this procedure is not long enough to cause any long-term damage, and the myocardium resumes normal function soon after. Angina is also believed to induce stunning. Jeroudi et al36 found that patients with angina experienced wall motion abnormalities that persisted for close to 24 hours, even after flow had been normalized and chest pain subsided. The implications of this study are somewhat uncertain because the delay of improvement can be linked to other factors, such as short-lived ischemic episodes that occur persistently, but sporadically, or myocardial hibernation.37 Also, it has been shown that the systolic function of patients who have been treated for acute myocardial infarction with thrombolytic therapy does not normalize until several days afterward. This delay in the improvement of myocardial function is strongly suggestive of myocardial stunning. Stress-induced ischemia is also believed to result in stunning. Ambrosio et al38 have demonstrated that after an exercise-induced ischemic episode, myocardial function remains depressed even after termination of exercise and normalization of perfusion levels. Stunning can be a dire consequence of cardiac surgery because the stunning may involve the entire left and right ventricles. This type of postoperative stunning, which may occur after cardiopulmonary bypass, may be alleviated by the use of perioperative inotropes.
Although there is no unified view of the pathogenesis of myocardial stunning, mainly because of species differences, the two main trigger mechanisms seem to be Ca2+ and oxygen radicals.
Although the seminal description of stunning was made in a large mammalian model,35 the Ca2+ hypothesis was largely developed in rodent models. During stunning in rodents, transient Ca2+ overload activates the Ca2+-dependent protease calpain I, which degrades, affects the phosphorylation state, and induces covalent modifications of myofilaments, such as troponin I (TnI).39 As a result, there is a decreased responsiveness to Ca2+, manifested by a decrease in the maximal force and a relative insensitivity to extracellular Ca2+ concentration. However, these data obtained in rodents differ radically from what is observed in larger mammals. As shown by Kim et al40 in a pig model of myocardial stunning where intracellular Ca2+ handling in myocytes from stunned and nonischemic regions were compared in the same heart, the impaired contractile function observed in vivo was also present in isolated myocytes in vitro. In this study, both Ca2+ transients and L-type Ca2+ current density were decreased in stunned myocytes, which indicates that the mechanisms of contractile dysfunction in the pig involve impaired Ca2+
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


