CONGENITAL HEART DISEASE IN THE ADULT
A little over a hundred years ago, Sir William Osler, in his classic textbook The Principles and Practice of Medicine (New York, Appleton & Co, 1892, pp 659–663), devoted only five pages to “Congenital Affections of the Heart,” with the first sentence declaring, that “[t]hese [disorders] have only limited clinical interest, as in a large proportion of cases the anomaly is not compatible with life, and in others nothing can be done to remedy the defect or even to relieve symptoms.” Fortunately, in the intervening century, considerable progress has been made in understanding the basis for these disorders and their effective treatment.
The most common birth defects are cardiovascular in origin. These malformations are due to complex multifactorial genetic and environmental causes, but recognized chromosomal aberrations and mutations of single genes account for <10% of all cardiac malformations. Congenital heart disease (CHD) complicates ~1% of all live births in the general population—about 40,000 births/year—but occurs more frequently in the offspring (about 4–5%) of women with CHD. Owing to the remarkable surgical advances over the last 60 years, >90% of afflicted neonates and children now reach adulthood; women with CHD may now frequently successfully bear children after competent repairs. As such, the population with CHD is steadily increasing. Women with aortic disease (e.g., aortic coarctation or Marfan’s syndrome) risk aortic dissection. Patients with cyanotic heart disease, pulmonary hypertension, or Marfan’s syndrome with a dilated aortic root generally should not become pregnant; those with correctable lesions should be counseled about the risks of pregnancy with an uncorrected malformation versus repair and later pregnancy.
More than 1 million adults with operated or unoperated CHD live in the United States today and, thus, outnumber the 800,000 children with CHD. Because true surgical cures are rare, and all repairs—be they palliative or corrective—may leave residua, sequelae, or complications, most require some degree of lifetime expert surveillance. The anatomic and physiologic changes in the heart and circulation due to any specific CHD lesion are not static but, rather, progress from prenatal life to adulthood. Malformations that are benign or escape detection in childhood may become clinically significant in the adult. For example, a functionally normal congenitally bicuspid aortic valve may thicken and calcify with time, resulting in significant aortic stenosis; a well-tolerated left-to-right shunt of an atrial septal defect (ASD) may result in cardiac decompensation or pulmonary hypertension only after the fourth to fifth decade.
CARDIAC DEVELOPMENT (ALSO SEE CHAPTER 1)
CHD is generally the result of aberrant embryonic development of a normal structure or failure of such a structure to progress beyond an early stage of embryonic or fetal development. This brief section serves to introduce the reader to normal development so that defects may be better understood; by necessity, it is not exhaustive. Cardiogenesis is a finely tuned process with transcriptional control of a complex group of regulatory proteins that activate or inhibit their gene targets in a location- and time-dependent manner. At about 3 weeks of embryonic development, two cardiac cords form and become canalized; at that point, the primordial cardiac tube develops from two sources (cardiac crescent or the first heart field, pharyngeal mesoderm or the second heart field); by 21 days, these fuse into a single cardiac tube beginning at the cranial end. The cardiac tube then elongates and develops discrete constrictions with the following segments from caudal to cranial location: sinus venosus receives the umbilical, vitelline, and common cardinal veins: atrium, ventricle, bulbus cordis, truncus arteriosus, aortic sac, and the aortic arches. The cardiac tube is fixed at the sinus venosus and arterial ends.
Subsequently, in the next few weeks, differential growth of cells causes the tube to elongate and loop as an “S” with the bulboventricular portion moving rightward and the atrium and sinus venosus moving posterior to the ventricle. The primitive atrium and ventricle communicate via the atrioventricular canal from which the endocardial cushion develops into two parts (ventrally and dorsally). The cushions fuse and divide the atrioventricular canal into two atrioventricular inlets and also migrate to help form the ventricular septum. The primitive atrium is divided first by a septum primum membrane, which grows down from the superior wall to the cushions; as this fusion occurs, the midportion resorbs in the center forming the ostium secundum. Rightward of the septum primum, a second septum secundum membrane grows down from the ventral-cranial wall toward—but not reaching—the cushions, and covering most, but not all, of the ostium secundum, resulting in a flap of the foramen ovale. The primitive ventricle is partitioned by a finely tuned set of events. The interventricular septum grows up toward the cushions, and the cushions form an upper inlet septum; between the two portions is a hole called the interventricular foramen. The left and right ventricles begin to develop side by side, and the atria and their respective inlet valves align over their ventricles. Finally, these two parts of the septum fuse with the bulboventricular ridges, which, once having septated the truncus arteriosus, extend into the ventricle. The bulbocordis divides into a subaortic portion as the muscular conus resorbs, while the subpulmonary section has elongation of its muscular conus. Spiral division of the common truncus arteriosus rotates and aligns the pulmonary artery and aortic portions over their respective outflow tracts, the aortic valve moving posterior over the left ventricle (LV) outflow tract and the pulmonary valve moving anterior over the right ventricle (RV) outflow tract, with a wraparound relationship of the two great arteries.
Early on, the venous systems are bilateral and symmetric and enter 2 horns of the sinus venosus. Ultimately, except for the coronary sinus, most of the left-sided portions and the left sinus–venosus horn regress and the systemic venous system empties into the right horn via the inferior and superior vena cavae. The pulmonary venous system, initially connecting to the systemic venous system, develops as buds from the developing lungs fuse together in the pulmonary venous confluence at which point the connection to the systemic system regresses. Simultaneously, a projection from the back wall of the left atrium (the common pulmonary vein) grows posteriorly to merge with the confluence, which then becomes a part of the posterior left atrial wall.
The truncus arteriosus and aortic sac initially develop six paired symmetric arches, which curve posteriorly and become the paired dorsal aortae. The detailed description of the selective regression of some of the arches is not presented in this chapter. In brief summary, this process results in the development of arch 3 as the internal carotid arteries, left arch 4 as the aortic arch and right subclavian artery, and part of arch 6 as the patent ductus arteriosus. The two dorsal thoracic aortae fuse in the abdomen with persistence of the left dorsal aorta.
SPECIFIC CARDIAC DEFECTS
Tables 19-1, 19-2, and 19-3 list CHD malformations as simple, intermediate, or complex. Simple defects generally are single lesions with a shunt or a valvular malformation. Intermediate defects may have two or more simple defects. Complex defects generally have components of an intermediate defect plus more complex cardiac and vascular anatomy, often with cyanosis, and frequently with transposition complexes. The goal of these tables is to suggest when cardiology consultation or advanced CHD specialty care is needed. Patients with complex CHD (which includes most “named” surgeries that usually involve complex CHD) should virtually always be managed in conjunction with an experienced specialty adult CHD center. Patients with intermediate lesions should have an initial consultation and subsequent occasional intermittent follow-up with a cardiologist. Patients with simple lesions often may be managed by a well-informed internist or general cardiologist, although consultation with a specifically trained adult congenital cardiologist is occasionally advisable.
TABLE 19-1
SIMPLE ADULT CONGENITAL HEART DISEASE
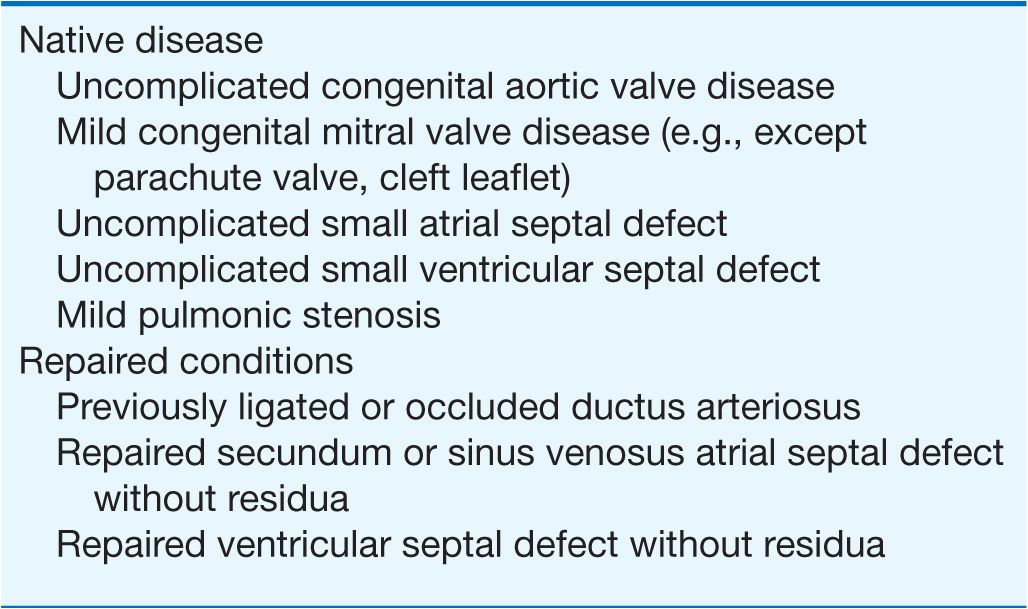
TABLE 19-2
INTERMEDIATE COMPLEXITY CONGENITAL HEART DISEASE

ATRIAL SEPTAL DEFECT
ASD is a common cardiac anomaly that may be first encountered in the adult and occurs more frequently in females. Sinus venosus ASD occurs high in the atrial septum near the entry of the superior vena cava into the right atrium and is associated frequently with anomalous pulmonary venous connection from the right lung to the superior vena cava or right atrium. Ostium primum ASDs lie adjacent to the atrioventricular valves, either of which may be deformed and regurgitant. Ostium primum ASDs are common in Down syndrome; the more complex atrioventricular septal defects with a common atrioventricular valve and a posterior defect of the basal portion of the interventricular septum are more typical of this chromosomal defect. The most common ostium secundum ASD involves the fossa ovalis and is midseptal in location; this should not be confused with a patent foramen ovale.
TABLE 19-3
COMPLEX ADULT CONGENITAL HEART DISEASE

Anatomic obliteration of the foramen ovale ordinarily follows its functional closure soon after birth, but residual “probe patency” is a common normal variant; ASD denotes a true deficiency of the atrial septum and implies functional and anatomic patency. The magnitude of the left-to-right shunt depends on the ASD size, ventricular diastolic properties, and the relative impedance in the pulmonary and systemic circulations. The left-to-right shunt causes diastolic overloading of the right ventricle and increased pulmonary blood flow. Patients with ASD are usually asymptomatic in early life, although there may be some physical underdevelopment and an increased tendency for respiratory infections; cardiorespiratory symptoms occur in many older patients. Beyond the fourth decade, a significant number of patients develop atrial arrhythmias, pulmonary arterial hypertension, bidirectional and then right-to-left shunting of blood, and right heart failure. Patients exposed to the chronic environmental hypoxemia of high altitude tend to develop pulmonary hypertension at younger ages. In older patients, left-to-right shunting across the ASD increases as progressive systemic hypertension and/or coronary artery disease (CAD) result in reduced compliance of the left ventricle.
Physical examination
Examination usually reveals a prominent RV impulse and palpable pulmonary artery pulsation. The first heart sound is normal or split, with accentuation of the tricuspid valve closure sound. Increased flow across the pulmonic valve is responsible for a mid-systolic pulmonary outflow murmur. The second heart sound is widely split and is relatively fixed in relation to respiration. A mid-diastolic rumbling murmur, loudest at the fourth intercostal space and along the left sternal border, reflects increased flow across the tricuspid valve. In ostium primum ASD, an apical holosystolic murmur indicates associated mitral or tricuspid regurgitation or a ventricular septal defect (VSD).
These findings are altered when increased pulmonary vascular resistance causes diminution of the left-to-right shunt. Both the pulmonary outflow and tricuspid inflow murmurs decrease in intensity, the pulmonic component of the second heart sound and a systolic ejection sound are accentuated, the two components of the second heart sound may fuse, and a diastolic murmur of pulmonic regurgitation appears. Cyanosis and clubbing accompany the development of a right-to-left shunt (see “Ventricular Septal Defect” later). In adults with an ASD and atrial fibrillation, the physical findings may be confused with mitral stenosis with pulmonary hypertension because the tricuspid diastolic flow murmur and widely split second heart sound may be mistakenly thought to represent the diastolic murmur of mitral stenosis and the mitral “opening snap,” respectively.
Electrocardiogram
In ostium secundum ASD, electrocardiogram (ECG) usually shows right-axis deviation and an rSr′ pattern in the right precordial leads representing enlargement of the RV outflow tract. An ectopic atrial pacemaker or first-degree heart block may occur with the sinus venous ASD. In ostium primum ASD, the RV conduction defect is accompanied by left superior axis deviation and counterclockwise rotation of the frontal plane QRS loop. Varying degrees of RV and right atrial (RA) enlargement or hypertrophy may occur with each type of defect, depending on the height of the pulmonary artery pressure. Chest x-ray shows an enlarged right atrium and right ventricle, and pulmonary artery and its branches; increased pulmonary vascular markings of left-to-right shunt vascularity will diminish if pulmonary vascular disease develops.
Echocardiogram
Echocardiography reveals pulmonary arterial and RV and RA dilatation with abnormal (paradoxical) ventricular septal motion in the presence of a significant right heart volume overload. The ASD may be visualized directly by two-dimensional imaging, color-flow imaging, or echo-contrast. In most institutions, two-dimensional echo-cardiography and Doppler examination have supplanted cardiac catheterization. Transesophageal echocardiography is indicated if the transthoracic echocardiogram is ambiguous, which is often the case with sinus venosus defects, or during catheter device closure (Fig. 19-1). Cardiac catheterization is performed if inconsistencies exist in the clinical data, if significant pulmonary hypertension or associated malformations are suspected, or if CAD is a possibility.
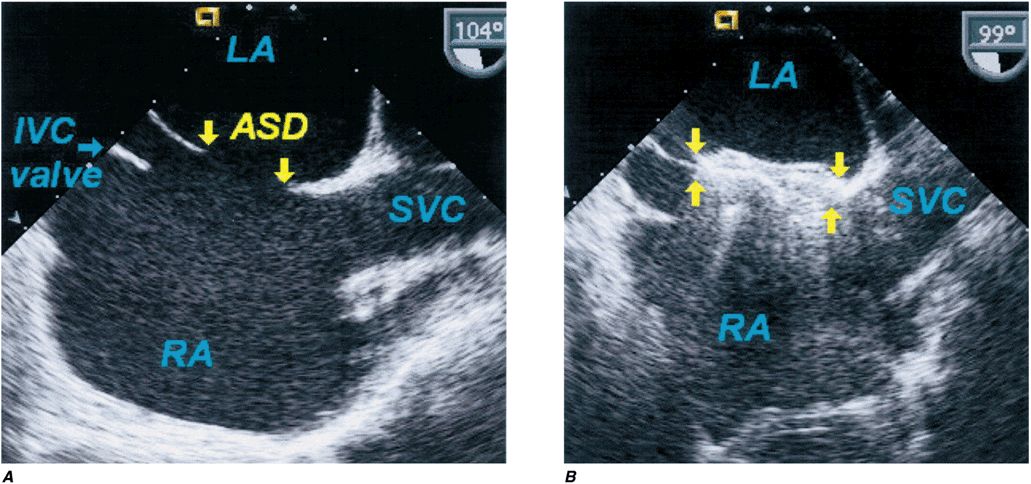
FIGURE 19-1
Secundum atrial septal defect. Transesophageal echocardiogram of secundum ASD and device closure. A. The atrial septal defect (ASD) between the left atrium (LA) and right atrium (RA) is shown. B. A percutaneous catheter–delivered device has occluded the defect. IVC, inferior vena cava; SVC, superior vena cava.
TREATMENT Atrial Septal Defect
Operative repair, usually with a patch of pericardium or of prosthetic material or percutaneous transcatheter device closure, if the ASD is of an appropriate size and shape, should be advised for all patients with uncomplicated secundum ASD with significant left-to-right shunting, i.e., pulmonary-to-systemic flow ratios ≥2:1. Excellent results may be anticipated, at low risk, even in patients >40 years, in the absence of severe pulmonary hypertension. In ostium primum ASD, cleft mitral valves may require repair in addition to patch closure of the ASD. Closure should not be carried out in patients with small defects and trivial left-to-right shunts or in those with severe pulmonary vascular disease without a significant left-to-right shunt.
Patients with sinus venosus or ostium secundum ASDs rarely die before the fifth decade. During the fifth and sixth decades, the incidence of progressive symptoms, often leading to severe disability, increases substantially. Medical management should include prompt treatment of respiratory tract infections; antiarrhythmic medications for atrial fibrillation or supraventricular tachycardia; and the usual measures for hypertension, coronary disease, or heart failure (Chap. 17), if these complications occur. The risk of infective endocarditis is quite low unless the defect is complicated by valvular regurgitation or has recently been repaired with a patch or device (Chap. 25).
Ventricular septal defect
A VSD is one of the most common of all cardiac birth defects, either as isolated defects or as a component of a combination of anomalies. The VSD is usually single and situated in the membranous or midmuscular portion of the septum. The functional disturbance depends on its size and on the status of the pulmonary vascular bed. Only small- or moderate-size VSDs are seen initially in adulthood, as most patients with an isolated large VSD come to medical or surgical attention early in life.
A wide spectrum exists in the natural history of VSD, ranging from spontaneous closure to congestive cardiac failure and death in infancy. Included within this spectrum are the possible development of pulmonary vascular obstruction, RV outflow tract obstruction, aortic regurgitation, or infective endocarditis. Spontaneous closure is more common in patients born with a small VSD, which occurs in early childhood in most. The pulmonary vascular bed is often a principal determinant of the clinical manifestations and course of a given VSD and feasibility of surgical repair. Increased pulmonary arterial pressure results from increased pulmonary blood flow and/or resistance, the latter usually the result of obstructive, obliterative structural changes within the pulmonary vascular bed. It is important to quantitate and compare pulmonary-to-systemic flows and resistances in patients with severe pulmonary hypertension. The term Eisenmenger’s syndrome is applied to patients with a large communication between the two circulations at the aortopulmonary, ventricular, or atrial levels and bidirectional or predominantly right-to-left shunts because of high resistance and obstructive pulmonary hypertension.
Patients with large VSDs and pulmonary hypertension are at greatest risk for developing pulmonary vascular obstruction. Large VSDs should be corrected surgically early in life when pulmonary vascular disease is still reversible or not yet developed. In patients with Eisenmenger syndrome, symptoms in adult life consist of exertional dyspnea, chest pain, syncope, and hemoptysis. The right-to-left shunt leads to cyanosis, clubbing, and erythrocytosis (see later). The degree to which pulmonary vascular resistance is elevated before operation is a critical factor determining prognosis. If the pulmonary vascular resistance is one-third or less of the systemic value, progression of pulmonary vascular disease after operation is unusual; however, if a moderate to severe increase in pulmonary vascular resistance exists preoperatively, either no change or a progression of pulmonary vascular disease is common postoperatively. Pregnancy is contraindicated in Eisenmenger syndrome. The mother’s health is most at risk if she has a cardiovascular lesion associated with pulmonary vascular disease and pulmonary hypertension (e.g., Eisenmenger physiology or mitral stenosis) or LV outflow tract obstruction (e.g., aortic stenosis), but she is also at risk of death with any malformation that may cause heart failure or a hemodynamically important arrhythmia. The fetus is most at risk with maternal cyanosis, heart failure, or pulmonary hypertension.
RV outflow tract obstruction develops in ~5–10% of patients who present in infancy with a moderate to large left-to-right shunt. With time, as subvalvular RV outflow tract obstruction progresses, the findings in these patients whose VSD remains sizable begin to resemble more closely those of the cyanotic tetralogy of Fallot. In ~5% of patients, aortic valve regurgitation results from insufficient cusp tissue or prolapse of the cusp through the interventricular defect; the aortic regurgitation then complicates and dominates the clinical course. Two-dimensional echocardiography with spectral and color Doppler examination defines the number and location of defects in the ventricular septum and associated anomalies and the hemodynamic physiology of the defect(s). Hemodynamic and angiographic study may be occasionally required to assess the status of the pulmonary vascular bed and clarify details of the altered anatomy.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


