Essentials of Genetic Disorders
Genetic factors play a significant role in all cardiovascular disorders (see also Chap. 10). Genetic defects are responsible for malformations of the heart and blood vessels, which account for the largest number of human birth defects. The estimated incidence of congenital heart disease is approximately 1% of all live births.1 The prevalence is estimated to be 10-fold higher among stillbirths.2 Genetic defects are responsible for familial cardiovascular disorders, such as cardiomyopathies and the long QT (LQT) syndrome, as well as nonfamilial sporadic forms. Evidence is now becoming available indicating genetic predisposition for common complex phenotypes, such as atherosclerosis and hypertension. Molecular genetics in conjunction with cytogenetics provide the opportunity to decipher the genetic basis and pathogenesis of cardiovascular diseases. Given the rapid pace of genetic discoveries, it is expected that genetic diagnosis and screening will become incorporated into standard practice in the near future. It is thus imperative that cardiologists understand the basis for genetic disorders and the medical and ethical implications of genetics.
All hereditary information is transmitted through DNA, a linear polymer composed of purine (adenine, guanine) and pyrimidine (cytosine, thymine) bases. The gene is the basic hereditary unit. It consists of a distinct fragment of DNA, which encodes a specific polypeptide (protein). There are approximately 30,000 genes in the human genome.3 Each individual has two copies of each gene, called alleles. The genes are localized in a linear sequence along 23 pairs of chromosomes, including 22 pairs of autosomes (chromosomes 1 to 22) and 1 pair of sex chromosomes, X and Y. Females have two X chromosomes, whereas males carry one X and one Y chromosome. Each parent contributes one of each chromosome pair (the members of the pair are referred to as homologous chromosomes) and thus one copy of each gene. The site at which a gene is located on a particular chromosome is referred to as the genetic locus. A given gene always resides at the same genetic locus on a particular chromosome, so the loci on homologous chromosomes are identical. However, alleles residing at these loci may be identical or different, leading to homozygous (identical alleles) and heterozygous (two different alleles present at the locus) states.
The genetic information is encoded by the linear sequence of the four bases of the DNA. Translation of this information into protein is through a translational code passed on through messenger ribonucleic acid (mRNA). Each unit of three bases, referred to as a codon, encodes a specific amino acid. The transcribed mRNA serves as the template that determines the sequence of the amino acids in the resulting polypeptide. Both autosomal alleles are usually transcribed into mRNA and translated into protein. However, expression of a gene can be restricted to specific cells and organs or regulated during a developmental stage because of regulation by cell- and tissue-specific transcription factors. In cells that carry two X chromosomes, only one X is active and the other X is silent after early embryogenesis.
In general, DNA nucleotide sequences remain stable during transmission to offspring. Nonetheless, occasional base sequence changes do occur, which are referred to as mutations. Mutations represent stable, heritable alterations in DNA. Somatic mutations, however, are not heritable. A number of mutagenic factors’such as environmental agents, radiation, chemicals, and errors by the DNA synthetic and editing enzymes—can induce mutations. Mutations can involve a visible alteration at the level of the chromosome (chromosomal abnormalities), which can result in the deletion or translocation of a portion of the chromosome, whereby several genes are often eliminated or altered. In contrast, mutations can be restricted to minor alterations in the DNA sequence, which vary from the substitution of a single nucleotide to that of the deletion or addition of multiple nucleotides. Thus hereditary and congenital diseases are conventionally classified into three broad categories: (1) chromosomal abnormalities, (2) single-gene or monogenic disorders, and (3) polygenic disorders or complex traits.
Each human cell has two copies of each chromosome (diploids) and each chromosome has two arms, referred to as the long, or “q,” and the short, or “p,” arms. The arms of the chromosomes meet at a primary constriction referred to as the centromere. Mutations typically occur during meiosis when chromosomes separate. Mutations can involve large deletions, duplications, translocations, rearrangements, and aneuploidy (too few or too many chromosomes). Chromosomal abnormalities are relatively common during embryonic life and lead to spontaneous abortion, often during the first trimester of pregnancy. However, a significant number of fetuses with chromosomal abnormalities survive. Chromosomal aberrations, numerical or structural, occur in approximately 1 in 150 liveborn infants.4 Most diseases caused by chromosomal abnormalities are detected in neonates or infants because involvement of many genes causes phenotypes that are easily diagnosed on physical examination. Chromosomal abnormalities often lead to structural heart defects and are found in 5% to 13% of liveborn children with congenital heart disease.1
The usual cause for gain of a chromosome is nondisjunction because of failure of a homologous pair of chromosomes to separate during meiosis. When an additional copy of the chromosome is added during fertilization, three copies of the same chromosome (or only one copy) are found in the new zygote instead of the chromosome pair. Two of the most common chromosomal disorders causing heart disease in the adult, namely Down syndrome (trisomy 21) and Turner syndrome (XO), are both commonly caused by nondisjunction. Chromosomal rearrangements occur when a chromosome breaks and rejoins within itself incorrectly, which can result in an inversion of the genetic material. Inversion occurs when a chromosome breaks at two points and the intermediate segment reunites in inverted orientation. Typically, there is no apparent phenotype in persons carrying an inversion, but their offspring may have severe abnormalities due to the disruption in chromosome pairing that can take place during meiosis. Isochromes are formed when two short or long arms join with loss of the other arm. Chromosomal translations occur when breaks arise in two chromosomes that are reunited after exchange of segments. Chromosome duplications or gains of chromosomal material may also be associated with phenotypic abnormality, but most commonly, they cause no obvious aberration.
Chromosome deletions are large deletions (equal to or greater than 106 base pairs) that commonly lead to loss of a large amount of DNA and loss or disruption of multiple genes. Consequently, a series of phenotypes in a single individual may be present as a result of interruptions in a series of genes within the loci of a single chromosome.
Each human genome also contains a large number of structural variations that include insertions, deletions, duplications, and rearrangement.5 The size of the structural variations could vary from 2 base pairs to several million base pairs. Insertion or deletion structural variations that change the copy number of the genes are commonly referred to as copy number variants (CNVs). Structural variants or CNVs are relatively common in the population but do not necessarily cause a gross phenotype, as typically observed in chromosomal abnormalities. However, structural variations including CNVs play significant roles in susceptibility to complex phenotypes, such as developmental abnormalities associated with congenital heart defects and neurologic disorders.
A single-gene disorder is an inherited disease that can be caused by a mutation in a single gene. Single-gene disorders show a mendelian pattern of inheritance. They are classified as autosomal dominant, autosomal recessive, or X-linked (dominant or recessive). The majority of monogenic diseases exhibit an autosomal-dominant mode of inheritance. Therefore, in a given family, approximately half of the members are affected. Monogenic disorders with an autosomal-recessive inheritance are caused by mutations in both copies of the gene. Therefore, in a given family only 25% of the offspring exhibit the phenotype, 50% carry the mutation, and 25% are normal. In X-linked inheritance, males exhibit the disease and females are usually free of the phenotype but carry the mutation. However, if the mutation involves a major protein, the effect of the mutation may be dominant and females can exhibit the clinical phenotype. In diseases caused by mitochondrial DNA mutations, inheritance is from the mother (no male-to-male transmission), because mitochondrial DNA is predominantly inherited from the ovum.
Only a fraction of cardiovascular disorders are monogenic. The DNA mutation gives rise to a change in the corresponding amino acids of the encoded protein and exerts its deleterious effects via functional alterations. A change in even one amino acid located in a critical domain of the protein can enhance the function (gain-of-function mutation) or impair the function (loss-of-function mutation), with a concomitant change in the phenotype. On average, a mutation occurs every 106 cell divisions or once every 200,000 years. Only mutations occurring in the gametes are transmitted.
In single-gene disorders, although the presence of the causal mutations is necessary for the development of the disease, other factors also affect the phenotypic expression of the disease. Modifier genes (the genetic background of the affected subjects) and the environmental factors are major determinants of phenotypic expression of a single-gene disorder.
Polygenic disorders such as coronary artery disease and related traits are caused by the interaction of genetic variants and nongenetic factors. Therefore, in this setting, the presence of a single variant is not sufficient to cause a disease, nor will its absence prevent development of the disease. Essentially, all of the common disorders have a genetic predisposition due to genetic variants that occur more frequently than that of rare single-gene disorders. Polygenic disorders account for the majority of the cardiovascular diseases, including atherosclerosis, hypertension, obesity, and diabetes mellitus. In polygenic diseases, multiple genes predispose to the condition. The mutations responsible for genetic predisposition in polygenic disorders usually involve single nucleotides distributed throughout the human genome with a frequency of approximately 1 per 1000 base pairs (bp).6,7 These single nucleotides imparting changes to the DNA sequence are referred to as single-nucleotide polymorphisms (SNPs). The effects of SNPs could confer protection against or susceptibility toward a complex phenotype and may be located in exons, introns, or intergenic regions.8 Technology made available in 2005 provided the opportunity to perform genome-wide association studies, which has led to the mapping of hundreds of loci predisposing to polygenic diseases, including coronary artery disease (discussed later).
Most human diseases exhibit genetic heterogeneity, defined as being caused by different genes and mutations causing the same phenotype. The heterogeneity may arise from multiple mutations in one gene (allele heterogeneity) or in two or more genes (locus heterogeneity). Within any one family, however, typically there is one causal gene and mutation in all affected members; uncommonly there are two different causal mutations or genes transmitted for the same disease. A good example is familial hypertrophic cardiomyopathy (HCM), which involves more than a dozen different genes (locus heterogeneity) with multiple mutations in each (allelic heterogeneity). In less than 10% of the cases, HCM is caused by double mutations.
Mutations can involve a microscopically visible alteration, such as deletion or translocation of a portion of the chromosome (chromosomal abnormalities), or a minute change in the DNA sequence, such as alteration of one purine or pyrimidine base. Mutations involving only a single nucleotide are known as point mutations and are responsible for ≥70% of all adult single-gene disorders. A point mutation may be a substitution of one nucleotide for another, changing the amino acid sequence (missense mutation), or it may change from encoding an amino acid to become a stop codon, which will truncate the protein (truncated or nonsense mutation), or it may eliminate a stop codon so the protein is elongated (elongated mutant). Finally, it may change the codon without changing the amino acid sequence (synonymous mutation). All genes during transcription and translation are read from 5′ to 3′ orientation, with each triplet of bases (codon) coding for a specific amino acid. If a nucleotide is deleted (deletion) or an additional nucleotide is inserted (insertion), it will shift the reading frame. The resulting protein would be entirely different (frameshift mutation) and usually nonfunctional. If a purine nucleotide is substituted for a pyrimidine or vice versa, the mutation is referred to as a transversion. If purine or pyrimidine substitutes for another purine or pyrimidine, respectively, it is called a transition. Other mutations may result from the deletion or addition of several nucleotides. In one form of myotonic dystrophy, for example, a triplet repeat of several thousand nucleotides in length is inserted into the 3′ end of the gene. Another type of mutation is known as a gene conversion, where two genes interact and part of the nucleotide sequence of one gene becomes incorporated into that of the other. Mutations in genes exert their deleterious effects via a structural alteration of the protein that has functional consequences, as noted.
The percentage of individuals within a family who have inherited the causal mutation and have one or more features of the disease is referred to as the penetrance. Penetrance is an all-or-none phenomenon. Any manifestation, however minute, indicates that the gene has penetrance in that individual. Nonpenetrance refers to lack of any observable phenotype. This feature is to be distinguished from expressivity, which refers to the variable nature of the clinical phenotype, such as the severity. Thus, by definition, to have expressivity, the trait must be penetrant. Numerous genetic and environmental factors can affect expression of a gene, making it nearly impossible to determine which factor is most important in a specific individual or disease. Table 82–1 shows these factors.
| 1. Causal genes and mutations |
| 2. Modifier genes (genetic background) |
| 3. Age |
| 4. Sex |
| 5. Exogenous or environmental factors such as exercise or diet |
| 6. Maternal factors |
| 7. Epigenetic alterations (such as DNA methylation) |
| 8. Posttranscriptional and posttranslation modifications |
| 9. Gene–gene (epistasis) and gene–environmental interactions |
| 10. MicroRNAs |
Inherited disorders caused by a single abnormal gene are transmitted to offspring in a predictable fashion, termed mendelian transmission. As previously noted, each individual has two copies of each gene, referred to as alleles, one transmitted from each parent. Per Mendel’s first law, each of the two alleles, located on separate chromosomes, segregates independently and is transmitted unchanged to offspring. Thus the chance of inheriting the mother’s allele versus the father’s is 50%. Mendel’s second law states that genes on the same chromosome also assert themselves independently through the process of crossover between segments of chromosomes (discussed below). The greater the distance between two loci, the more likely they are to be separated during genetic transmission. Mutant genes located on any of the 22 autosomal pairs or the 2 sex chromosomes may produce phenotypes inherited by simple patterns classified as autosomal (dominant or recessive) or X-linked, respectively. The terms dominant inheritance and recessive inheritance refer to characteristics of the phenotype. Dominant inheritance implies that a person with one copy of a mutant allele and one copy of the normal allele develops the phenotype associated with the mutant allele. Recessive traits, on the other hand, require both alleles to be mutant in order to produce a phenotype.
Dominant disorders are those exhibiting a phenotype in heterozygous individuals, as noted. Males and females are equally affected, and offspring of an affected heterozygote have a 50% chance of inheriting the mutant allele. In a sporadic case, the mutation occurs de novo and in one of the germ lines of parents (typically sperm). By definition, it is absent in the somatic cells of parents. Autosomal-dominant inheritance can be misdiagnosed as sporadic if there is low expressivity in the phenotypically normal parent carrying the mutant allele or if extramarital paternity has occurred. Table 82–2 lists the features characteristic of autosomal-dominant inheritance.
| A. Autosomal-dominant transmission |
| 1. Each affected individual has an affected parent unless the disease occurred because of a new mutation or the heterozygous parent has low expressivity. |
| 2. Equal proportions (ie, 50-50) of normal and affected offspring are likely to be born to an affected individual. |
| 3. Normal children of an affected individual bear only normal offspring. |
| 4. Equal proportions of males and females are affected. |
| 5. Both sexes are equally likely to transmit the abnormal allele to male and female offspring, and male-to-male transmission occurs. |
| 6. Vertical transmission through successive generations occurs. |
| 7. Delayed age of onset. |
| 8. Variable clinical expression. |
| B. Autosomal-recessive transmission |
| 1. Parents are clinically normal (in alternate generations) but genetically are heterozygotes. |
| 2. Alternate generations are affected, with no vertical transmission. |
| 3. Both sexes are affected with equal frequency. |
| 4. Each offspring of heterozygous carriers has a 25% chance of being affected, a 50% chance of being an unaffected carrier, and a 25% chance of inheriting only normal alleles. |
| C. X-linked transmission |
| 1. No male-to-male transmission. |
| 2. All daughters of affected males are carriers. |
| 3. Sons of carrier females have a 50% risk of being affected, and daughters have a 50% chance of being carriers. |
| 4. Affected homozygous females occur only when an affected male and carrier female have children. |
| 5. The pedigree pattern in X-linked recessive traits tends to be oblique because of the occurrence of the trait in the sons of normal carriers but not in the sisters of affected males (ie, uncles and nephews affected). |
| D. Mitochondrial transmission |
| 1. Equal frequency and severity of disease for each sex. |
| 2. Transmission through females only, with offspring of affected males being unaffected. |
| 3. All offspring of affected females may be affected. |
| 4. Extreme variability of expression of disease within a family (may include apparent nonpenetrance). |
| 5. Phenotype may be age-dependent. |
| 6. Organ mosaicism is common. |
Autosomal-recessive phenotypes are clinically apparent when the patient carries two mutant alleles (ie, is homozygous) at the locus responsible for the disease (Fig. 82–1). The disease-causing gene is found on 1 of the 22 autosomes. Thus both males and females are equally affected. Clinical uniformity is typical, and disease onset generally occurs early in life. Recessive disorders are more commonly diagnosed in childhood than are dominant diseases. On average, only one in four children (25%) will be affected (see Table 82–2).
Figure 82–1.

This typical set of pedigrees outlines the usual inheritance patterns for autosomal-dominant and autosomal-recessive traits, X-linked inheritance, and mitochondrial inheritance. Squares signify males; circles signify females. Filled-in circles and squares are affected females and males, respectively.
X-linked inherited disorders are caused by defects in genes located on the X chromosome. Because females have 2 X chromosomes, they may carry either one mutant allele (heterozygote) or two mutant alleles (homozygote). The trait may therefore display dominant or recessive expression. Males have a single X chromosome (and one Y chromosome). Consequently, a male is expected to display the full syndrome whenever he inherits the abnormal gene from his mother. Hence the terms X-linked dominant and X-linked recessive apply only to the expression of the gene in females. Because a male must pass on his Y chromosome to all male offspring, he cannot pass on a mutant X allele to his sons. Therefore, no male-to-male transmission in X-linked disorders can occur. On the other hand, a male must contribute his one X chromosome to all daughters (see Fig. 82–1). All females receiving a mutant X chromosome are known as carriers, and those who become affected clinically with the disease are known as manifesting female carriers. Table 82–2 lists the characteristic features of X-linked inheritance. Examples of X-linked disorders of the heart include X-linked cardiomyopathy, Barth syndrome, and Duchenne, Becker, and Emery-Dreifuss muscular dystrophies.
Spermatocytes contribute few or no mitochondria to the zygote. The entire mitochondrial DNA in an embryo is derived from the mitochondria already present in the cytoplasm of the oocyte. Thus phenotypes caused by mitochondrial DNA mutations demonstrate only maternal inheritance (see Fig. 82–1). Table 82–2 lists the characteristic features of mitochondrial inheritance.
Overview of Gene Mapping and Mutation Detection
Until the 1980s, identification of a disease-causing gene without knowing the causal protein was nearly impossible. For the majority of diseases, neither the defect nor the protein was known. Technical advances that made chromosomal mapping feasible include (1) computerized linkage analysis, (2) development of highly informative DNA markers spanning the entire genome, and (3) detection of markers by polymerase chain reaction (PCR). The 46 chromosomes of the human genome contain 3.2 billion bp of DNA. To locate a particular gene, one must first map the chromosomal locus, which requires knowledge of certain chromosomal landmarks, referred to as DNA markers. A DNA marker is a polymorphic sequence of DNA with a known chromosomal position, which can be detected by analyzing an individual’s DNA (discussed in detail below). Initial genetic linkage studies were performed using short-tandem repeat (STR) DNA markers. STR markers are available that span each chromosome at intervals of not more than 4 million base pairs (Mbp) on all chromosomes (a set of approximately 800 markers). In recent years, SNPs have been used for genetic linkage as well. The typical SNP linkage panel contains several thousands SNPs that are positioned throughout the genome, with an average distance of approximately 0.5 Mbp. Genetic distance is measured in terms of centimorgans (cM), named after the geneticist T.H. Morgan. One cM approximates 1 Mbp. DNA markers like genes have two alleles in a given individual and are transmitted to offspring according to Mendel’s law, with the individual being heterozygous or homozygous for that marker. If a marker is homozygous, it is not informative for genetic linkage. When all of the markers are placed together on each chromosome and the genetic distance between them is estimated, a genetic map is produced. Several maps of more than 5000 highly informative DNA markers that span the entire genome have been developed.9
Identification of a particular locus is made possible by showing that the causal gene of interest is in close proximity to a DNA marker on the same chromosome, a method referred to as genetic linkage analysis. A fundamental requirement for linkage analysis is a family in which the disease of interest is transmitted to offspring over at least two and preferably three generations. At least six affected individuals are required for analyzing cosegregation of DNA markers with inheritance of the disease, although a larger number of affected individuals is preferable.
The homologous pairs of chromosomes are assorted, and one from each parent is transmitted to the offspring by chance. Each gene, allele, or marker is transmitted independently. Thus the odds of any two genes (or a marker and a gene) being coinherited is 50% (chance alone). Even genes on the same chromosome are transmitted independently by the mechanism of crossover between homologous chromosomes (Fig. 82–2), unless they are in close physical proximity to each other. In the latter case, they cosegregate together. Homologous recombination provides for continual mixing of the genes during every meiosis. It is the predominant reason why no two individuals have the same genotype for all DNA markers unless they are identical twins. Before meiosis, the two homologous chromosomes come together and form bridges (chiasmata) such that segments of equal proportion are exchanged between them, giving rise to crossover between homologous regions of various genes. In genetic parlance, crossing over is referred to as recombination. The loci occupy the same chromosomal position on the homologous chromosome on which they are combined as they had on their original homologous chromosome. There is no net loss of chromosomal material or genes, but crossover leads to a constant intermixing of the chromosomes such that no two offspring will ever be identical. Crossovers occur only between homologous chromosomes. On average, there are 33 crossovers between homologous chromosome pairs per meiosis.
Figure 82–2.
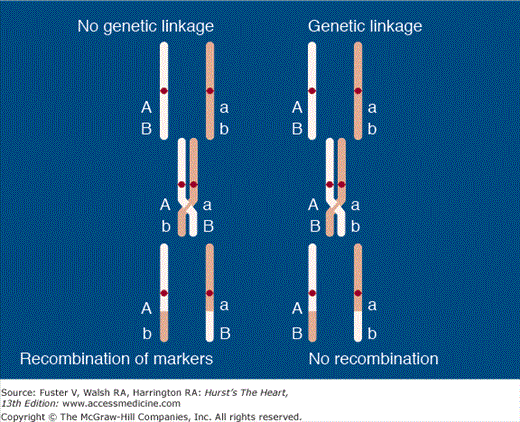
Linkage analysis. Loci A (disease locus) and B (DNA marker locus) are located in close proximity, with minimal chance of crossover between them. Thus even when crossover occurs between homologous segments of chromosomes during meiosis, A and B loci cosegregate together and thus are considered genetically linked.
The most important part of an evaluation for genetic disease is the family history. First, this may give clues to the diagnosis of a particular phenotype and inheritance patterns within an individual family. For instance, an individual’s ethnic background may suggest the need for specific types of genetic screening, such as for hemoglobinopathies in individuals of African or Mediterranean ancestry or for Tay-Sachs disease in individuals of eastern European (Ashkenazi) Jewish ancestry. The individual with the medical problem who brought the family to the attention of the physician is referred to as the proband or propositus (proposita for females) or index case. Information should generally be collected on all individuals who are first-, second-, or third-degree relatives of the proband. First-degree relatives of the proband are the parents and children. Second-degree relatives are aunts and uncles, grandparents, and grandchildren of the proband. Third-degree relatives are first cousins, great aunts and uncles, great-grandparents, and great-grandchildren. A pedigree chart (see Fig. 82–1) is then generated. This information should include medical problems and pregnancies. If relatives are deceased, the age at death and the cause of death should be recorded. With a pedigree chart and specific family information, general questions are asked, including whether other family members have the same or similar problems. Information about various types of birth defects, mental retardation, early infant deaths, miscarriages, stillbirths, or other diseases or handicaps in the family is sought. With some disorders, there may be a variability of a particular condition (ie, clinical heterogeneity), even within a family. For example, with a possible diagnosis of HCM, one should ask about premature death or syncope. A pregnancy history may provide information to support a possible teratogenic exposure. The date of the last menstrual period, whether the pregnancy was planned, whether contraception was used immediately before pregnancy, the time when the pregnancy was recognized, and when the mother sought prenatal care should be noted. Problems during the pregnancy’such as bleeding, spotting, cramping, fevers, rashes, or illnesses; drug exposures (both prescribed and nonprescribed), alcohol intake, or “recreational” drug use; and exposures to potent chemicals in the workplace or while involved in various hobbies’should be explored. Pregnancy and family histories can then be used in conjunction with the findings on physical examination to derive a potential etiologic diagnosis and to plan for further diagnostic studies. The term etiologic diagnosis should suggest whether a specific cardiac defect is familial (by family history), genetic but not familial (sporadic), teratogenic (by pregnancy history), or multifactorial. Prognosis and recurrence risk are linked strongly to an accurate diagnosis and its probable etiology. In sum, accurate phenotypic characterization is essential for all genetic studies.
Despite the independent assortment of chromosomes and genes during meiosis, genes (alleles) on two or more loci are often coinherited because they are so close together that a chiasmatic bridge does not form between them. Two loci coinherited more than 50% of the time are considered genetically linked. To map the chromosomal locus responsible for a causal gene, DNA markers that are evenly distributed across the chromosomes are selected. DNA is collected from all members of a family (normal and affected) and genotyped for the selected markers. If a DNA marker is coinherited with the phenotype in the affected individuals, the chromosomal locus where the DNA marker resides is in close physical proximity to the locus of the causal gene. This is referred to as genetic linkage between the disease (causal gene) and the DNA marker. Figure 82–2 illustrates the concept of linkage analysis. Shown in the left panel is an illustration of genetic linkage between a locus for a DNA marker and that of a disease that is inherited in a mendelian dominant fashion. The locus, designated with an “A,” carries the allele responsible for the disease. The corresponding locus, “a,” on the homologous chromosome has the allele that codes for the same protein but has not undergone a mutation and is thus the normal allele. The loci designated “B” and “b” represents alleles of a DNA marker of known location that has nothing to do with the disease. In the right panel, the disease and the marker loci are so close that they tend to be coinherited within the family. In contrast, in the left panel, the “A” and “b” loci are so far apart that recombination and crossover occur between the two markers, and thus they segregate independently. The calculation necessary to prove definitively that genetic linkage does or does not exist between a DNA marker and a disease-related locus is sophisticated and requires advanced computer programs. The odds for and against linkage are calculated. Linkage exists if the odds in favor of linkage are at least 1000:1. Commonly, the logarithm of the odds, referred to as the LOD score (log of the odds), is used, and a LOD score of ≥3 indicates linkage. A LOD score of −2 (ie, 102 or 100:1 odds against linkage) excludes the linkage. The likelihood of two genes being separated by recombination increases in proportion to the distance between them. The distance between a marker and a disease-causing gene when genetically linked is quite variable and may be anywhere from 1 to 50 Mbp but is usually within 1 to 10 Mbp. Thus the inherent resolution of genetic linkage analysis is not better than 1 Mbp.
It is possible on the basis of linkage analysis alone to construct a chromosomal map of all of the DNA markers, with the distance between the various markers estimated in centimorgans. This is a complex calculation derived from the number of recombinations between the DNA markers during meioses. The recombination frequency between two markers, two genes, or a gene and a marker is the ratio of the number of crossover events to the total number of meioses. The lower the recombination frequency between the locus of a DNA marker and that of a disease-causing gene, then the closer those two are in physical distance on the chromosome. However, despite the close physical proximity of the loci of the DNA marker and the disease-causing gene, recombination still may occur. The extent to which recombination does occur reflects roughly the physical distance between the two loci. The recombination fraction (or theta) is used to develop a means of estimating the genetic distance (in centimorgans) between genetically linked loci. A recombination frequency or crossover of 1% between two loci, whether occupied by two genes or one gene and a DNA marker, reflects a physical distance of approximately 1 cM between them. For a marker and a gene separated by 1 cM, this means the chance of a crossover between them during meiosis is only 1%; thus the chance of their being coinherited is 99%. This is a statistically derived genetic map, however, and the distances are only approximate.
Once the chromosomal location of a gene has been mapped, the first technique in attempting to identify the gene is referred to as the positional candidate gene approach. There are approximately 230,000 genes in the genome, however, more than 100,000 expressed sequenced tags (ESTs) have been mapped. These ESTs are unique DNA sequences of 100 to 200 bp, each of which is claimed to represent a portion of the expressed sequences of a gene. These genes and ESTs are available through a worldwide network of databases.
Known genes or ESTs in the mapped chromosomal region are amplified as candidate genes that contain the causative mutation that segregates with the disease. If none of the candidate genes in the region are shown to have a mutation that cosegregates with the disease, it may be necessary to clone the region. This approach is referred to as positional cloning, so named because a region is cloned knowing only its position relative to the genetically linked DNA markers. Positional cloning is usually unnecessary, as most genes in the human genome have been mapped and identified. However, if attempted, it is necessary to reduce the region (containing the gene) to ≥1 cM. It is often necessary to expand the family with the hope of finding crossovers such that DNA markers common to all affected would span only a short distance (<1 cM). The cloned genes or PCR-amplified DNA is then analyzed, commonly by direct sequencing, for the presence of the mutation. To strengthen the causality, the mutation must be shown to cosegregate with the disease and not with the unaffected members in the family. In addition, it is crucial to show that the variant is absent in large number of normal individuals with the same ethnic background, and hence it is not a polymorphism. Finally, to establish the causality, in vivo and in vitro functional studies are necessary. Table 82–3 summarizes the approach to chromosomal mapping of hereditary diseases by linkage analysis and subsequent isolation of the gene. Mapping of genes predisposing to polygenic disorders10-12 is discussed under Genetics of Coronary Artery Disease.
| 1. Identification of a family with a familial disease. |
| 2. Collection of clinical data from the family. |
| 3. Clinical assessment to provide an accurate diagnosis of the disease using a consistent and objective criterion to separate normal individuals from those affected and from those who are indeterminate or unknown. |
| 4. Collection of blood samples for immediate DNA analysis and development of lymphoblastoid cell lines for a renewable source of DNA. |
| 5. Development of a family pedigree. |
| 6. DNA analysis for markers of known chromosomal loci that span the human genome in an attempt to find a marker locus linked to the disease. |
| 7. Identification of the gene. |
| 8. Identification of mutation(s) causing the disease. |
| 9. Demonstration of a causal relationship between the mutant gene and the disease. |
| 10. Development of a convenient test to screen for the mutation. |
Genetic Counseling Principles
Genetic counseling should provide information about the diagnosis, possible etiology, and prognosis of a disease. In addition, psychosocial issues, reproductive options, and the availability of prenatal diagnosis should be discussed. Genetic counseling should be nondirective, providing information in a nonjudgmental, unbiased manner. The family should then be able to make decisions based on medical information in the context of their religious, moral, cultural, and social backgrounds and their financial situation. Although a genetic counselor may occasionally feel frustrated with a specific couple’s decision, an effective counselor does not let personal biases interfere with the counseling role. Conflicts leading to major ethical issues and disputes may arise, however, and may be particularly apparent regarding issues of nonpaternity, sex selection, pregnancy termination, and selective nontreatment of malformed infants. Couples have many potential reproductive options, but not all may be acceptable religiously or culturally. Nevertheless, potential options should be mentioned in a sensitive manner. A common misunderstanding among families in genetic counseling is the issue of prenatal diagnosis and its relationship to abortion. Prenatal diagnosis does not imply that a parent should or would terminate the pregnancy. In many circumstances the information from prenatal diagnosis may help to reassure a couple that their risk of having another handicapped child is, in fact, much lower than expected. Conversely, if defects are found, the subspecialist may use more diagnostic approaches to make rational decisions about medical management of the infant before or immediately after delivery.
The accelerated pace of progress in gene discovery, molecular medicine, and molecular diagnostics has begun to allow for improved genetic counseling and portends the possibility of future genetic therapy. As knowledge about the genetic basis of disease grows, however, so does the potential for discriminatory health insurance policies to exclude individuals who are at risk for an illness or to charge prohibitively high rates on the basis of predetermined illness. For this reason planners of the Human Genome Project recognized the need to protect individuals who volunteer for genetic study as well as those diagnosed by molecular methods in the future. Also for this reason, the National Institutes of Health–Department of Energy Working Group on Ethical, Legal, and Social Implications of the Human Genome Project was developed. After 13 years of debate in Congress, on May 21, 2008, President Bush signed into law the Genetic Information Nondiscrimination Act. The bill protects Americans against discrimination based on genetic information for hiring or insurance.
Cardiovascular Abnormalities Caused by Chromosomal Defects
Table 82–4 lists chromosomal defects that cause cardiovascular abnormalities. The most common chromosomal defects are described briefly below.
| Chromosome Defects | Syndromes | Cardiac Phenotype |
|---|---|---|
| 45X | Turner syndrome | Coarctation of the aorta, ASD, aortic stenosis |
| Trisomy 5 | Interrupted aortic arch | |
| Trisomy 13 | Patau syndrome | CHD, VSD |
| Trisomy 18 | Edwards syndrome | CHD, VSD |
| Partial trisomy 20q | Dextrocardia | |
| Trisomy 21 | Down syndrome | CHD, ASD, VSD, PDA |
| Trisomy 22 | VSD | |
| Partial tetrasomy 22 | Schmid-Fraccaro syndrome | CHD |
| Anomalous pulmonary venous return | ||
| Deletion 4p | Wolf-Hirschhorn syndrome | CHD |
| Deletion 7q11.23 | Williams syndrome | CHD, supravalvular aortic stenosis, hypertension, MVP |
| Deletion paternal 15q11 | Prader-Willi syndrome | CHD |
| Deletion 17p | Miller Dieker syndrome | CHD, ASD |
| Deletion 22q11 | CATCH-22, DiGeorge, and velocardiofacial syndromes | CHD |
| Rearrangement 5p15.1-3 | Cri du chat | CHD |
| Recombination chromosome 8 | San Luis Valley syndrome | Tetralogy of Fallot |
Turner syndrome is characterized by a constellation of findings that result from partial or complete monosomy of the X chromosome.13 It is the most common chromosomal abnormality in females, with an incidence of 1 per 2500 to 3000 liveborn girls, which corresponds to approximately 2 million cases worldwide.14 It is characterized by cardiovascular anomalies, short stature, low-set ears, excess nuchal skin, broad chest with widely spaced nipples, peripheral lymphedema, and ovarian dysgenesis. Cardiac abnormalities are common, with a prevalence estimated to be between 23% and 40%.15 The most common cardiovascular abnormalities are bicuspid aortic valve, which is present in 10% to 20%, and coarctation of aorta, present in 10% of adult cases. The prevalence of these abnormalities is higher in children. Less common cardiovascular anomalies include aortic stenosis, systemic hypertension, mitral valve prolapse, conduction defects, partial anomalous venous drainage, and ventricular septal defect (VSD). Aortic dilatation and dissection, partly because of concomitant hypertension, also occur (see also Chap. 106). Women with Turner syndrome are more susceptible to aortic aneurysms and ischemic heart disease.14,15 Patients with Turner syndrome should undergo periodic cardiovascular evaluation, including 12-lead ECG and echocardiography.
Turner syndrome is caused by complete or partial absence of an X chromosome. The most common karyotype is monosomy X (45,X).13 Approximately 5% to 10% of cases have duplication of the long arm of 1 X (46,X,i[Xq]) and the rest have mosaicism.13 The pathogenesis of Turner syndrome is not fully understood. It likely entails haploinsufficiency of genes (located on the X chromosome) that, under normal conditions, escape inactivation. Inactivation of 1 copy of the X chromosome during early embryogenesis is partial, and several genes escape inactivation. Specific genes that account for cardiovascular phenotype in Turner syndrome are unknown. SHOX (short stature homeobox-containing gene) or PHOG (pseudoautosomal homeobox-containing osteogenic gene), which encode two isoforms of a homeodomain protein, are considered responsible for the short stature in Turner syndrome.14 Zinc finger protein X and zinc finger protein Y genes (ZFX/ZFY), which are involved in sex determination, are also candidate genes for Turner syndrome.16
Down syndrome, or trisomy 21, is a major cause of mental retardation and congenital heart disease, with a characteristic set of facial and physical features. The incidence of Down syndrome is approximately 1 in 700 livebirths, affecting more than 350,000 individuals in the United States alone.17 The risk of having a live-born infant with Down syndrome increases with maternal age. It is estimated at 1 in 1000 at age 30 years and 10-fold higher at age 45 years.18 The recurrence rate in the offspring is approximately 1%. Clinical manifestations include congenital anomalies of the heart and gastrointestinal tract, epicanthal folds, flattened facial profile, small and rounded ears, up-slanted palpebral fissures, excess nuchal skin, and brachycephaly. An increased risk of leukemia, immune system defects, and an Alzheimer-like dementia are associated with Down syndrome. Cardiac abnormalities are present in approximately half of the cases.19,20 The most common cardiac abnormalities are atrioventricular canal defect and isolated VSD, which occur in 45% and 35% of cases, respectively.19,20 Isolated secundum atrial septal defect (ASD) is present in 8% and tetralogy of Fallot in 5% of cases (see also Chap. 9).19
Down syndrome is caused by trisomy 21. It is full trisomy in 95%, chromosomal translocation in 2%, and mosaic in 3%.21,22 The vast majority of errors in meiosis leading to trisomy 21 are of maternal origin and occur during the first meiosis in two-thirds and during second meiosis in one-fifth of the cases. The exact causal genes responsible for the cardiovascular defects are unknown. However, four Down syndrome critical regions (DSCRs) have been mapped.22-25 DSCR1 encompasses an area of approximately 5 million bp and approximately 20 genes.22 DNA markers in this region are associated with mental retardation, and most of the facial features of the syndrome.22 Among the candidate genes is DSCR1, which is abundantly expressed in the heart and brain.26 It is a candidate for cardiac anomalies and mental retardation.26 High-resolution map of the phenotype of congenital heart defects in patients with the Down syndrome locus restricts the region to a 1.7-Mbp interval, which contains 10 genes. Among the candidates in this region is DSCAM, which encodes Down syndrome cell adhesion molecule. It is the only gene in this region that is expressed in the heart.27 The pathogenesis of Down syndrome is unknown. Down syndrome is considered a contiguous gene syndrome.17 It is expected to involve increased expression of multiple contiguous genes. Overexpression of DSCR1, a product of DSCR, is shown in the brains of patients with Down syndrome.28DSCR1 encodes calcipressin 1, which functions through direct binding and inhibition of calcineurin A, the catalytic subunit of the Ca2+/calmodulin-dependent serine threonine protein phosphatase (PP2B).29 Calcineurin dephosphorylates nuclear factor of activated T cells (NFAT), which leads to its nuclear localization and induction of gene expression.29 Inhibition of calcineurin by DSCR1 is expected to increase levels of phosphorylated NFAT and reduce nuclear localization of NFAT and NFAT-mediated gene expression. Inhibition of calcineurin by DSCR1 may be one of the multiple mechanisms involved in the pathogenesis of Down syndrome.30
Edwards syndrome, or trisomy 18, is the second most common trisomy, with a prevalence of approximately 1 in 4000 to 8000 livebirths.31 The majority of the infants die within a couple of weeks and approximately 10% survive more than a year.32 The syndrome is characterized by anomalies of the heart and microcephaly with a prominent occiput, a narrow forehead, low-set and malformed ears, micrognathia, clefting of the lip and palate, clenched hand with overlapping digits, rocker-bottom feet, and various hernias.21 Cardiovascular anomalies are present in approximately 90% of cases. They include VSD, ASD, patent ductus arteriosus (PDA), pulmonary stenosis, tetralogy of Fallot, transposition of the great arteries, bicuspid aortic valve, dysplastic valves, and coarctation of the aorta.19,21 Full trisomy occurs in more than 85%, chromosomal translocation in 3%, and mosaicism in 5% of cases.21 The causal gene(s) for the cardiovascular anomalies remains unknown.
Patau syndrome, or trisomy 13, is a rare disorder with an incidence of 1 per 5000 to 1 in 20,000 livebirths and a high early mortality.21 Approximately 50% of affected infants die within the first month and 85% within first year of life.21 Patau syndrome is characterized by cardiac, urogenital, craniofacial, and central nervous system anomalies. Specific anomalies include microcephaly with sloping forehead, microphthalmia, cleft lip and palate, overlapping fingers with postaxial polydactyly, and renal abnormalities, including polycystic kidney disease. Cardiac abnormalities are present in approximately 80% of the cases. They include VSD, ASD, PDA, pulmonary stenosis, coarctation of the aorta, dextrocardia, and truncus arteriosus.19,21
Patau syndrome is caused by nondisjunction of chromosome 13 during meiosis in the vast majority of cases and rarely by translocation. Five percent of the cases are mosaic. The causal genes for cardiovascular anomalies in trisomy 13 are unknown.
DiGeorge and velocardiofacial syndromes are autosomal-dominant congenital anomalies caused by hemizygous microdeletion of a large segment of the long arm of chromosome 22 (22q11). The deletion leads to anomalies of multiple organs, including the heart and facial bones. The prevalence is approximately 1 in 4000, accounting for approximately 15% of all congenital heart defects.32,33 The term CATCH-22 denotes cardiac, abnormal facies, thymic hypoplasia, cleft palate, hypocalcemia (as a result of parathyroid hypoplasia), and the 22nd chromosome. Cardiac anomalies are present in approximately two-thirds of cases. A diverse array of congenital heart defects, including tetralogy of Fallot, interrupted aortic arch, truncus arteriosus, and PDA, have been described. Tetralogy of Fallot is the most common abnormality.32 Patients with velocardiofacial syndromes exhibit craniofacial anomalies, cleft palate, and a variety of cardiac abnormalities, such as aortic arch anomalies, tetralogy of Fallot, and VSD. Cardiac valves and the myocardium are usually spared.
DiGeorge syndrome is caused by microdeletion of approximately 3 Mbp of DNA encompassing approximately 30 genes.32 Genetic analysis in mouse and mutation analysis of the candidate genes in the region have led to identification of mutations in TBX1, which encodes a T-box transcription factor.34 TBX1 is critical for embryogenesis of aortic and pulmonary outflow tracts. Loss-of-function mutations in TBX1 result in haploinsufficiency. The downstream target genes of TBX1 and the pathways involved in the pathogenesis of cardiac phenotype are mostly unknown.
Another candidate gene is UFD1L, which encodes a protein involved in degradation of ubiquitinated proteins. It is expressed during the embryogenesis of cell lines typically associated with DiGeorge syndrome. Similarly, UFD1L is expressed in association with the conotruncus and the fourth embryologic aortic arch. A large deletion in human transcription factor UFD1L in a single patient with a phenotype similar to that of DiGeorge syndrome has been identified.35 Deletion of Ufd1L in mice produced some of the typical cardiac phenotypes that result from defective development of the fourth branchial arch.36 However, several other studies have excluded mutations in UFD1L in patients with DiGeorge syndrome.37 Other genes, such as ZNF74, which encodes a zinc-finger transcription factor, also have been implicated in the pathogenesis of DiGeorge syndrome.38 However, the causal role remains to be established.
Genetic Basis of Specific Congenital Heart Diseases
A significant number of congenital heart diseases occur in isolation and are not part of complex phenotypes as observed in chromosomal abnormalities. Recently, the causal genes for several congenital heart diseases have been identified. Preliminary studies depict a common theme in the pathogenesis of isolated congenital heart defects, which implicate deficiency of several transcriptional factors that regulate cardiac gene expression during embryogenesis. However, there is considerable phenotypic, locus, and allelic heterogeneity.
Supravalvular aortic stenosis39 is an autosomal-dominant disease characterized by discrete narrowing of the ascending aorta above the level of the sinus of Valsalva. It commonly occurs as a phenotype of Williams syndrome (or Williams-Beuren syndrome) in conjunction with mental retardation in some, and exceptional talents in others, hypercalcemia, characteristic facial appearance, and stenosis of other major arteries. The prevalence of supravalvular aortic stenosis is estimated to be 1 in 25,000 livebirths.
The gene responsible for supravalvular aortic stenosis was initially mapped to chromosome 7q11.23 and subsequently identified as ELN, encoding elastin.41 Almost all cases of isolated supravalvular aortic stenosis are caused by ELN mutations, which comprise a variety of point and deletion mutations.40 Mutations result in elastin deficiency, which in the vascular system leads to inelasticity of the vessel wall and subsequent fibrosis as a result of an altered stress–strain relation (elastin arteriopathy). Thus haploinsufficiency underlies the pathogenesis of supravalvular aortic stenosis.
Patients with Williams syndrome may exhibit additional cardiovascular phenotypes, including pulmonary arterial stenosis, aortic and mitral valve abnormalities, and tetralogy of Fallot.41 In 98% of cases of Williams syndrome, the deletion mutation includes 1.5 Mbp of DNA comprising ELN and another 20 contiguous genes. Contribution of these genes to pathogenesis of specific phenotypes in Williams syndrome remains unknown. The genetic basis of William-Beuren syndrome has been reviewed in detail in Schubert.42
ASD is among the most common congenital heart diseases, with an estimated incidence of 1 in 1000 livebirths.1 ASD is usually sporadic. However, familial ASD with an autosomal-dominant mode of inheritance has also been described.43,44 Individuals with ASD are commonly asymptomatic until the third or fourth decades. Common symptoms are palpitations, commonly caused by supraventricular arrhythmias, and symptoms associated with pulmonary hypertension and right-sided volume overload resulting in left-to-right shunt. Uncorrected ASD can lead to heart failure and premature death in the fourth or fifth decade of life.
The first gene identified for familial ASD is NKX2–5 (CSX1), which is the human homologue of Nkx2.5 in mouse and tinman in Drosophila melanogaster.45 The gene is located on 5q35 and encodes NKX2.5, a predominantly cardiac-specific transcription factor that regulates expression of several cardiac genes.46 A multiplicity of mutations has been described in patients with secundum ASD and conduction defects.45,47 Mutations often result in haploinsufficiency. Point mutations in the DNA-binding domain reduce the affinity of NKX2.5 for the promoter regions and hence decrease expression of cardiac-specific genes.48 The spectrum of clinical phenotypes caused by mutations in NKX2.5 extends beyond secundum ASD and comprises VSDs, tetralogy of Fallot, subvalvular aortic stenosis, pulmonary atresia, and others.49
The second causal gene for familial ASD with an autosomal-dominant mode of inheritance is GATA4 on chromosome 8p22-23.50 The mutations diminish DNA-binding affinity and transcriptional activity of GATA4 transcription factor and block its physical interaction with TBX5, another transcription factor involved in the pathogenesis of congenital heart disease.50
The third causal gene for familial ASD is MYH6, which is located in chromosome 14q12 and encodes myosin heavy chain 6.51 A missense mutation in MYH6 in a family with atrial fibrillation mapped to 14q12 locus has been identified.51 The MYH6 protein is expressed at high levels in atrial tissues and plays an important role in formation of interatrial septum.51
Holt-Oram syndrome is a rare autosomal-dominant inherited disorder characterized by anomalies of the heart and upper extremities, hence the name hand–heart syndrome (see also Chap. 84).52 The most common congenital heart defects are ASD and VSD, followed by conduction system abnormalities and atrial fibrillation.53 Less common cardiac abnormalities include truncus arteriosus, mitral valve defect, PDA, and tetralogy of Fallot.53 Anomalies of the upper limb vary from mild malformation of the carpal bones to phocomelia, but upper limb preaxial radial abnormalities are commonly present.53
Mutations in TBX5 on chromosome 12q24, which codes for transcription factor TBX5, are responsible for the cardiac and skeletal abnormalities in Holt-Oram syndrome.53,54 A number of mutations have been described, and most are nonsense, frameshift, or splice-junction abnormalities. The proposed molecular mechanism is haploinsufficiency, resulting in reduced expression level of TBX5. Haploinsufficiency because of truncation or frameshift mutations results in severe birth defects in the heart and hands, whereas point mutations predominantly affect either hand or heart development.55 Mutations in the 5′ end of the gene exhibit a preponderance of cardiac abnormalities with mild skeletal abnormalities, and those in the 3′ end lead to severe skeletal and mild cardiac abnormalities.
Ellis–van Creveld syndrome is an autosomal-recessive skeletal dysplasia, which is associated with congenital heart disease in the majority of cases. Skeletal anomalies include short limbs, short ribs, postaxial polydactyly, and dysplastic nails and teeth. ASD and common atrium are the typical cardiac anomalies present in two-thirds of the cases (see also Chap. 84).
The gene responsible for Ellis–van Creveld syndrome was mapped to chromosome 4p16.156 near an area proximal to the FGFR3 gene, which is known to cause hypochondroplasia and achondroplasia. Subsequently, splice donor, truncation, and missense mutations in a novel gene, EVC, were identified.57 Mutations in EVC account for approximately 20% of the cases of Ellis–van Creveld syndrome.58 Recently, mutations in a second gene, named EVC2, for Ellis–van Creveld syndrome, were identified.58 The pathogenesis of Ellis–van Creveld syndrome remains unknown.
PDA can occur as a sole cardiac anomaly or in conjunction with other congenital heart disease. Familial PDA with an autosomal-dominant inheritance has been described in patients with Char syndrome. Char syndrome is a congenital disease that was first described by Florence Char in 1978 and is characterized by a constellation of facial dysmorphism, fifth-finger middle phalangeal hypoplasia, and PDA. Variation of this syndrome is associated with bicuspid aortic valve, distinctive facial appearance, polydactyly, and fifth-finger clinodactyly. The predominant clinical features are those of PDA, which include symptoms and signs of left-heart failure and pulmonary hypertension.
The gene responsible for Char syndrome in two families was recently mapped to chromosome 6p12-21.59 Subsequently, mutations in TFAP2B, which encodes a neural crest-related helix-span-helix transcription factor, were identified.60 These findings suggest that Char syndrome results from derangement of neural crest-cell derivatives.60 A second locus for familial PDA has been mapped to a 3-cM interval on chromosome 12q24.61 The causal gene remains unknown.
Noonan syndrome is an uncommon autosomal-dominant disorder characterized by dysmorphic facial features, HCM, pulmonic stenosis, mental retardation, and bleeding disorders.62 LEOPARD syndrome (lentigines, electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonic stenosis, abnormal genitalia, retardation of growth, and deafness) is an allelic variant of the Noonan syndrome.62 Pulmonic stenosis and HCM are the primary cardiac phenotypes. Others include atrioventricular septal defects, aortic coarctation, ASD, mitral valve defects, PDA, and fibroelastosis. Noonan syndrome is also seen in conjunction with cardiofaciocutaneous syndrome and other congenital abnormalities, such as neurofibromatosis (see also Chap. 9).
Noonan syndrome is sporadic in half of the cases and autosomal-dominant in the other half. The gene responsible for autosomal-dominant Noonan and LEOPARD syndromes was mapped to chromosome 12q22 and subsequently identified as encoding protein-tyrosine-phosphatase, nonreceptor type 11 (PTPN11).63,64 With the exception of deletion of amino acid glycine 60, all mutations in PTPN11 are missense mutations.62 Most mutations are recurrent and localized to exons 3, 7, 8, and 13.62 The N308D mutation is the most common and accounts for approximately 25% of cases.62 Overall, mutations in PTPN11 are found in approximately two-thirds of cases of Noonan syndrome.62
Mutations are located in interacting portions of the amino-terminal src-homology 2 (N-SH2) and protein-tyrosine-phosphatase (PTP) domains.63,64 Both gain-of-function and dominant-negative mechanisms have been implicated in the pathogenesis of Noonan syndrome caused by PTPN11 mutations.
Mutations in KRAS, SOS1, and RAF1 also have been identified in patients with Noonan syndrome. Collectively, mutations in the above three genes account for approximately 25% of all Noonan syndrome cases.
Myxomas are the most common cardiac tumors and are generally sporadic. Myxomas are familial, with an autosomal-dominant mode of inheritance in approximately 10% of cases (see also Chap. 84).65 Familial myxoma commonly occurs as a part of Carney complex, with the constellation of cardiac myxoma, endocrine disorders, and skin pigmentation.66 LAMB (lentigines, atrial myxoma, mucocutaneous myxoma, and blue nevi) and NAME (nevi, atrial myxoma, myxoid neurofibromata, and ephelides) syndromes are considered variants of Carney complex. Atrial, ventricular, and skin myxomas; endocrine tumors and disorders such as Cushing syndrome; and skin lesions such as lentiginosis are part of the phenotypic expression of Carney complex. Clinical features of atrial myxoma may include fever, arthralgia, dyspnea, diastolic rumble, tumor plop, and systemic embolisms.
Carney complex exhibits locus heterogeneity, and at least two loci on chromosome 17q24 and 2p16 have been mapped.67,68 The majority of familial cardiac myxomas (Carney complex) are caused by mutations in the PRKRA1A gene on chromosome 17q24.69 It encodes the α-regulatory subunit of cyclic adenosine monophosphate (cAMP)–dependent protein kinase. Frameshift mutations in PRKRA1A result in haploinsufficiency, which suggests that the PRKRA1A functions as a tumor-suppressor gene. Recently, a missense mutation in the perinatal myosin heavy-chain gene (MYH8) was identified in members of a family with Carney complex and trismus-pseudocamptodactyly syndrome.70
Situs inversus is a reversal of the asymmetric anatomic position of visceral organs. In situs inversus totalis, all visceral organs are reversed in a mirror-image manner. It is part of the immotile cilia syndrome (primary ciliary dyskinesia). Kartagener syndrome is situs inversus, bronchiectasis, and male sterility. Most cases of situs inversus are sporadic. Autosomal-recessive, autosomal-dominant, and X-linked forms have been reported.
Situs inversus, as a component of immotile cilia syndrome, such as that in Kartagener syndrome, is caused by mutations in dyneins.71 Dyneins are large proteins with adenosine triphosphatase (ATPase) activity that interact with intermediary filaments to produce energy and motion. Mutations in dynein axonemal intermediate chain 1 (DNAI1) on chromosome 9p13-p21, dynein axonemal heavy chain 5 (DNAH5) on chromosome 5p, and dynein axonemal heavy chain type 11 (DNAH11) on chromosome 7p21 have been found in patients with primary ciliary dyskinesia (and situs inversus).71-73
Situs inversus has also been mapped to chromosome Xq26.2. Mutations in ZIC3, encoding a zinc-finger protein of the cerebellum, are associated with situs ambiguus in male and situs solitus or inversus in females.74 Other causal genes for right-left axis abnormality include CFC1 on chromosome 2, LEFTB (also known as LEFTY2) and ACVR2B, encoding activin receptor IIB.75,76
Alagille syndrome is an autosomal-dominant disorder characterized by anomalies of the right side of the heart and developmental abnormalities of eyes, skeleton, and kidney. Cardiac abnormalities are present in approximately 70% of cases; the most common is diffuse pulmonary artery stenosis. Others include hypoplastic pulmonary circulation, pulmonary atresia, tetralogy of Fallot, coarctation of aorta, secundum ASD, PDA, and VSD.77 The most common causal gene is Jagged-1 gene (JAG1), located on chromosome 20p12.78,79 Deletion or point mutations in JAG1 are found in approximately 90% of the patients with Alagille syndrome. JAG1 is a cell surface protein that is a ligand for the Notch receptor. The Notch intercellular signaling pathway mediates cell fate decisions during development. The proposed molecular mechanism is haploinsufficiency leading to defective cell adhesions. Recently, mutations in NOTCH2 were found in those who did not have JAG1 mutations.80 Collectively, the findings indicate that Alagille syndrome is disease of Notch signaling pathway.
Genetic Diseases of Cardiac Muscle
The term cardiomyopathy denotes an exclusive group of disorders in which the primary defect is in the myocardium, affecting cardiac myocyte structure and/or function. The primary defect, however, does not need to be exclusive to the heart. It can also involve other tissues and organs, as in cardiomyopathies arising from metabolic disorders and mitochondrial myopathies. Myocardial dysfunction can also occur because of systemic, infiltrative, toxic, and endocrine disorders; coronary atherosclerosis; and valvular pathologies. In such conditions, the primary defect is not in the myocardium. Thus myocardial involvement is considered secondary. In a sense, cardiac involvement in such disorders does not meet the pure definition of cardiomyopathy. In the broader definition, the Council on Clinical Cardiology, Heart Failure and Transplantation Committee of the American Heart Association provided the following definition for cardiomyopathies: “Cardiomyopathies are a heterogeneous group of diseases of the myocardium associated with mechanical and/or electrical dysfunction that usually (but not invariably) exhibit inappropriate ventricular hypertrophy or dilatation and are due to a variety of causes that frequently are genetic”81 (see Chap. 84).
Cardiomyopathies are classified according to their phenotypic characteristics. The common form groups are hypertrophic, dilated, restrictive, and arrhythmogenic right ventricular cardiomyopathy. Phenotypic classification, although clinically convenient and useful, does not sufficiently reflect the molecular and genetic basis of cardiomyopathies. Future classification of cardiomyopathies is expected to be based on our understanding of their molecular pathogenesis.
HCM is a relatively common autosomal-dominant disease diagnosed clinically by the presence of unexplained cardiac hypertrophy.82 Commonly, a left ventricular wall thickness of ≥13 mm, in the absence of hypertension or valvular heart disease, is used to define HCM. The prevalence of HCM is approximately 1 in 500 in young adults.83 It is likely higher in the elderly population because of age-dependent penetrance.
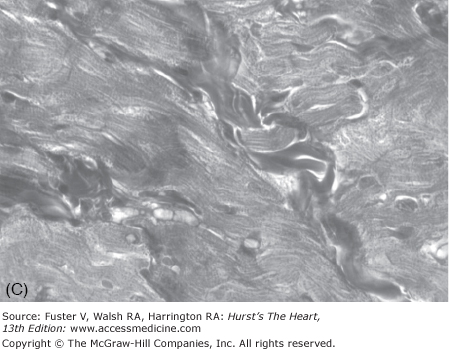
Cardiac hypertrophy, the clinical hallmark of HCM, is asymmetric in approximately two-thirds of cases, with predominant involvement of the interventricular septum (Fig. 82–3). Hence the term asymmetric septal hypertrophy is used to describe this condition. Rarely, hypertrophy is restricted to apex of the heart (apical HCM). Morphologically, the left ventricular cavity is small, and left ventricular ejection fraction, a measure of global systolic function, is increased. However, more sensitive indices of myocardial function show impaired contraction and relaxation.84 Diastolic function is commonly impaired, leading to an increased left ventricular end-diastolic pressure and thus, frequently, to symptoms of heart failure (see Chap. 84).


Patients with HCM exhibit protean clinical manifestations ranging from minimal or no symptoms to severe heart failure. The clinical manifestations often do not develop until the third or fourth decade of life, but the onset is variable. The majority of patients are asymptomatic or mildly symptomatic. Predominant symptoms include dyspnea, chest pain, palpitations, and/or syncope. Severe systolic heart failure is uncommon. It occurs in a small fraction of patients in whom the disease evolves into a dilated cardiomyopathy (DCM) phenotype.85 In contrast, cardiac diastolic function is usually impaired and left ventricular end-diastolic pressure is elevated. A dynamic left ventricular outflow is present in approximately 25% of patients. It could contribute to mitral regurgitations and symptoms of heart failure. Cardiac arrhythmias, in particular atrial fibrillation and nonsustained ventricular tachycardia, are relatively common and are associated with adverse clinical outcome.86,87 Wolff-Parkinson-White (WPW) syndrome is present in a small percentage of patients with HCM. Its presence suggests the possibility of a phenocopy, typically a glycogen storage disease.88-91
Syncope is a serious symptom. It is often a result of serious cardiac arrhythmias and associated with an increased risk of sudden cardiac death (SCD).92-94 HCM, although uncommon, is the most common cause of SCD in young, competitive athletes.95,96 It accounts for almost half of all cases of SCD in athletes younger than 35 years of age in the United States.95,96 SCD is often the first and tragic manifestation of HCM in young, apparently healthy individuals.95,96 Table 82–5 lists the factors associated with an increased risk of SCD.97 Overall, in the assessment of the risk of SCD, combination of all known risk factors should be considered.94 In the absence of major risk factors for SCD, HCM has a relatively benign course, with an estimated annual mortality of approximately 1% in the adult population.98-100 Apical HCM, characterized by giant T-wave inversion in the precordial leads on the electrocardiogram, is also a relatively benign disease.101,102
| Established risk factors |
| Prior episode of aborted SCD (sudden cardiac arrest) |
| Family history of SCD (more than one victim of SCD) |
| Causal mutations, including double mutations |
| Modifier genes (genetic background) |
| History of syncope |
| Severe cardiac hypertrophy |
| Sustained and repetitive nonsustained ventricular tachycardia |
| Less-established risk factors |
| Left ventricular outflow tract gradient |
| Histologic phenotypes (interstitial fibrosis and myocyte disarray) |
| Early onset of clinical manifestations (young age) |
| Abnormal blood pressure response to exercise |
| Presence of myocardial ischemia |
The pathologic hallmark of HCM is cardiac myocyte disarray.103 It is defined as malaligned, distorted, and often short and hypertrophic myocytes oriented in different directions (see Fig. 82–3). Myocyte disarray often comprises more than 20% of the ventricle, as opposed to <5% of the myocardium in the normal hearts.103-105 It is more prominent in the interventricular septum, but commonly is found throughout the myocardium.103,105 Other pathologic features of HCM include myocyte hypertrophy, interstitial fibrosis, thickening of media of intramural coronary arteries, and sometimes malpositioned mitral valve with elongated leaflets. Cardiac hypertrophy, interstitial fibrosis, and myocyte disarray are associated with the risk of SCD, mortality, and morbidity in patients with HCM.106-109 Other pathologic features of HCM include thickening of the media of intramural coronary arteries, abnormal positioning of the mitral valve apparatus, and elongated mitral valve leaflets.
HCM is a genetically heterogeneous disease with an autosomal-dominant mode of inheritance. Approximately two-thirds of patients have a family history of HCM.110-112 In the remainder, the disease is sporadic. Familial and sporadic cases both are caused by mutations in contractile sarcomeric proteins.109 In sporadic cases, mutations are de novo and could be transmitted to the offspring of the index cases.113,114 Because hypertrophy is a common response of the heart to all forms of injury or stimuli, a phenotype of hypertrophy in the absence of an increased external load could also occur because of mutations in nonsarcomeric proteins. As such, unexplained cardiac hypertrophy, which clinically denotes HCM, could also occur in storage disorders,115 metabolic disorders,116 mitochondrial diseases,117 and triplet repeat syndromes,118 as well as congenital heart diseases.63 Although the gross phenotype is similar, the pathogenesis of HCM caused by different classes of mutant proteins, at least in part, could differ. Therefore, such conditions are considered phenocopy (diseases mimicking HCM).
The pioneering works of Christine and Jonathan Seidman have led to elucidation of the molecular genetic basis of HCM. In 1990, an arginine-to-glutamine substitution at codon 403 (R403Q) in the β-myosin heavy chain (MHC) was identified as the first causal mutation.119 Since then, several hundred different mutations in more than a dozen genes encoding sarcomeric proteins have been identified (Table 82–6). Consequently, HCM (excluding phenocopy) is considered a disease of contractile sarcomeric proteins (Fig. 82–4).120 Systematic screening of sarcomeric genes suggests that mutations in MYHC and MYBPC3, which encode β-MHC and myosin-binding protein C (MBP-C), respectively, are the most common causes of human HCM, together accounting for approximately half of all cases.121-123 Mutations in TNNT2 and TNNI3, encoding cardiac troponin T and I, respectively, are relatively uncommon, each accounting for approximately 3% to 5% of the HCM cases.123-125 Thus mutations in MYH7, MYBPC3, TNNT2, and TNNI3 collectively account for approximately 60% of all HCM cases. A small fraction of HCM cases are caused by mutations in genes encoding α-tropomyosin (TPM1), titin (TTN), cardiac α-actin (ACTC), telethonin (TCAP), and essential and regulatory light chains (MYL3 and MYL2, respectively).120,126-131 Rare mutations in cardiac troponin C (TNNC1), α-MHC (MYH6), myosin light chain kinase (MYLK2), phospholamban (PLN), and caveolin 3 (CAV3) have been reported in patients with HCM.132-136 Finally, mutations in MYOZ2 and TCAP encoding Z disc proteins myozenin 2 (or calsarcin 1) and telethonin, respectively, are also uncommon causes of HCM 137,138 Overall, the causal genes and mutations for approximately two-thirds of HCM cases have been identified. The remainder are yet to be identified or are a result of genes inducing a phenocopy.
| Gene | Symbol | Locus | Frequency | Predominant Mutations |
|---|---|---|---|---|
| β-Myosin heavy chain | MYH7 | 14q12 | ∼25% | Missenses |
| Myosin binding protein-C | MYBPC3 | 11p11.2 | ∼25% | Splice-junction and insertion/deletion |
| Cardiac troponin T | TNNT2 | 1q32 | ∼3%-5% | Missenses |
| Cardiac troponin I | TNNI3 | 19p13.2 | ∼3%-5% | Missense and deletion |
| α-Tropomyosin | TPM1 | 15q22.1 | <3% | Missenses |
| Essential myosin light chain | MYL3 | 3p21.3 | <3% | Missenses |
| Regulatory myosin light chain | MYL2 | 12q23-24.3 | <3% | Missense and one truncation |
| Cardiac α-actin | ACTC | 15q11 | <3% | Missense mutations |
| Titin | TTN | 2q24.1 | <3% | Missense mutations |
| Telethonin (Tcap) | TCAP | 17q2 | Rare | Missense mutations |
| α-Myosin heavy chain | MYH6 | 14q1 | Rare | Missense and rearrangement mutations (association) |
| Cardiac troponin C | TNNC1 | 3p21.3-3p14.3 | Rare | Missense mutations (association) |
| Cardiac myosin light peptide kinase | MYLK2 | 20q13.3 | Rare | Point mutations (association) |
| Caveolin 3 | CAV3 | 3p25 | Rare | Point mutations (association) |
| Phospholamban | PLN | 6p22.1 | Rare | Point mutations (association) |
| Myozenin 2 | MYOZ2 | 4q26-q27 | 1:250 | Point mutations |
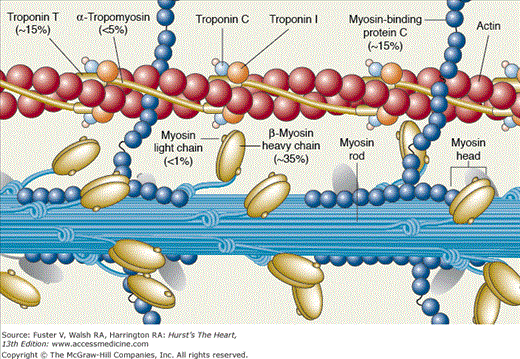
More than 100 different mutations in the β-MHC, a major component of thick filaments in sarcomeres, have been identified. The majority are missense mutations, localized in the globular head of the myosin molecule. Codons 403 and 719 are potential hotspots for mutations.139,140 Missense, deletion, and insertion/deletion mutations in the rod and tail regions have also been described but are uncommon.141-145 Overall, the frequency of each particular MYHC mutation is relatively low, and most mutations are private. Accordingly, a founder effect (sharing of a common ancestor) is uncommon.
Mutations in MYBPC3 account for approximately 25% of all HCM cases. Mutations are scattered throughout the gene without a particular predilection.122,146-149 Unlike mutations in MYHC, a significant proportion of MYBPC3 mutations are deletion/insertion or splice-junction mutations.122 Insertion/deletion mutations could result in frameshift and truncation of the MBP-C protein. The mutant protein harbors severe structural and functional defects or becomes degraded. The frequency of each particular mutation is relatively low, and a founder effect is uncommon.
Mutations in TNNT2 are relatively common causes of human HCM, accounting for approximately 3% to 5% of all cases.120,125,126,150,151 More than 20 mutations in TNNT2 have been described, and codon 92 is considered a hotspot for mutations.120,150,151 The majority of the mutations are missense, but deletion mutations that involve splice donor sites and could lead to truncated proteins also have been described.120
Mutations in TNNI3 are also relatively uncommon and estimated to account for approximately 3% to 5% of all HCM cases.123,124,152,153 Mutations in other components of thin and thick filaments are uncommon causes of HCM and collectively account for approximately 5% of all HCM cases. Mutations in other sarcomeric genes, namely TPM1,TNNC1, TTN,ACTC,MYL3, and MYL2, are very uncommon, and those in MYH6, myosin light chain kinase, SERCA2A, and phospholamban are rare.
A remarkable feature of HCM is the presence of considerable variability in its phenotypic expression, whether it is the degree of cardiac hypertrophy or the risk of SCD. The molecular basis of such variability is not fully known. It is probably partly because of the diversity of the causal genes and mutations, which impart a spectrum of functional and structural defects.154 In addition, the presence of multiple mutations, detected in a small fraction of patients, is associated with a severe phenotype.155,156 Environmental factors, such as competitive sports and exercise, could potentially contribute to the phenotypic expression of HCM.157,158 However, there are insufficient data to support their contributions to the phenotype.
The presence of considerable phenotypic variability among affected members of different families with identical causal mutations emphasizes the significance of the genetic background to phenotypic expression of HCM. Genes other than the causal genes that affect the phenotype are referred to as the modifier genes. Unlike the causal genes, modifier genes are neither necessary nor sufficient to cause HCM.159 However, they influence the severity of cardiac hypertrophy, risk of SCD, and expression of other cardiac phenotypes in HCM. DNA polymorphisms, including putatively functional SNPs (ie, SNPs located in the coding or regulatory regions or splice junctions) in genes involved in cardiac hypertrophy, are the prime candidates. The identity of the modifier genes for HCM and the magnitude of their effects remain largely unknown. Five modifier loci have been mapped, and several genes have been implicated.160,161 The effect sizes of the modifier loci are considerable, as the loci in homozygous form drastically influence expression of cardiac hypertrophy in patients with the causal mutation. A modifier locus also has been mapped in a genetically engineered mouse model.162 Functional variants of genes coding for the components of the renin-angiotensin-aldosterone system are the most extensively studied candidates. ACE, encoding angiotensin-I converting enzyme 1 (ACE-1), was the first gene implicated as a modifier of cardiac phenotype in human HCM.163 ACE-1 catalyzes conversion of angiotensin I to angiotensin II and inactivates bradykinin, both of which are biologically active agents with opposing effects on cardiac growth and cellular proliferation.164ACE contains more than 30 different polymorphisms, which contribute to interindividual variation in plasma, tissue, and cellular levels of ACE-1.165 The most commonly studied ACE polymorphism is an insertion (I) or deletion (D) of a 287-bp Alu repeat in intron 16. The I/D polymorphism, probably because of being in LD with other functional SNPs, is associated with variation in plasma, cellular, and tissue levels of ACE-1 in a codominant manner (DD > ID > II).165
ACE I/D polymorphism has been associated with the severity of cardiac hypertrophy and risk of SCD in HCM in most, but not all, studies.162-172 The DD genotype is more common in HCM families with a high incidence of SCD, as compared with those with a low incidence, and is associated with the severity of cardiac hypertrophy.163,166 The observed association is gene-dose dependent, consistent with the biologic effect of the I/D variants on plasma and tissue levels of ACE.166 An interaction between the modifying effect of the I/D genotypes and the underlying causal mutations also has been reported.167,172
Variants of endothelin-1 (EDN1), tumor necrosis factor-α (TNF-α), angiotensinogen (AGT), angiotensin II receptor 1 (AGTR1), and platelet-activating factor acetylhydrolase (PLA2G7) have been associated with the severity of the cardiac hypertrophy.173-175 The results, however, have been inconsistent, partly because of the small sample size of the studies, population characteristics, and presence of confounders that are common in SNP-association studies.176
Mutations in nonsarcomeric proteins can cause a phenotype grossly similar to HCM, referred to as a phenocopy.115 The distinction between true HCM and HCM phenocopy is important because the pathogenesis, as well as histologic phenotypes, of the two conditions differ. Table 82–7 provides a partial list of HCM phenocopy. The prevalence of HCM phenocopy is not precisely known. Given the prevalence of each particular HCM phenocopy, that phenocopy is expected to comprise approximately 5% to 10% of the cases with the clinical diagnosis of HCM.115 A prototypic example of HCM phenocopy is Fabry disease, an X-linked lysosomal storage disease.177,178 Fabry disease is present in approximately 3% of cases with the clinical diagnosis of HCM in adult population.179 The phenotype results from deficiency of α-galactosidase A (α-Gal A), also known as ceramide trihexosidase.180 The deficiency of the enzyme results in deposits of glycosphingolipids in multiple organs, including the heart. The causal gene is GLA on chromosome Xq22, which codes for lysosomal hydrolase α-Gal A protein.180 The phenotype is characterized by angiokeratoma, renal insufficiency, proteinuria, neuropathy, transient ischemic attack, stroke, anemia, corneal deposits, and cardiac hypertrophy.180 Cardiac hypertrophy, which is often indistinguishable from the true HCM, is associated with high QRS voltage, conduction defects, and cardiac arrhythmias. Other cardiac phenotypes include valvular regurgitation, coronary artery disease, myocardial infarction, and aortic annular dilatation.179,181 The disease predominantly affects males. Female carriers could exhibit a milder form.181 The diagnosis is established by measuring α-Gal A levels and activity in leukocytes. Fabry disease can be treated with enzyme replacement therapy using human α-Gal A (agalsidase α) or recombinant human α-Gal A (agalsidase β).177,178,182,183
| Gene | Gene Symbol | Chromosome | Frequency |
|---|---|---|---|
| Protein kinase A, γ subunit | PRKAG2 | 7q22-q31.1 | 1%-2% |
| α-Galactosidase A {2468} | GLA | Xq22 | 3% |
| Unconventional myosin 6 | MOY6 | 6q12 | Rare |
| Lysosome-associated membrane protein 2 | LAMP2 | Xq24 | 1%-2% |
| Mitochondrial genes | MTTG, MTTI | MtDNA | Rare |
| Frataxin (Friedreich ataxia) | FRDA | 9q13 | Rare |
| Myotonin protein kinase (Myotonic dystrophy) | DMPK, DMWD | 19q13 | Uncommon |
| Protein tyrosine phosphatase, nonreceptor type 11 | PTPN11 | 12q24 | Uncommon, higher in children |
Glycogen storage disease, caused by mutations in PRKAG2 gene, is another HCM phenocopy.88-91 Cardiac hypertrophy results predominantly from storage of glycogen in myocytes. The gene encodes the γ2 regulatory subunit of adenosine monophosphate (AMP)–activated protein kinase (AMPK), which is considered the energy biosensor of the cell. Mutations in PRKAG2 lead to cardiac hypertrophy, conduction defects, and WPW.88-90
HCM phenocopy also occurs in trinucleotide repeat syndromes, a group of genetic disorders caused by expansion of naturally occurring trinucleotide repeats.184 HCM phenocopy occurs in Friedreich ataxia, an autosomal-recessive neurodegenerative disease caused by expansion of GAA repeat sequences in the intron of FRDA.185
HCM phenocopy also occurs in patients with Noonan syndrome. The phenotype is characterized by dysmorphic facial features, pulmonic stenosis, mental retardation, bleeding disorders, and cardiac hypertrophy. LEOPARD syndrome is an allelic variant of Noonan syndrome. Noonan syndrome is an autosomal-dominant disorder caused by mutations in PTPN11, KRAS, SOS1, and RAF1 in approximately three-quarters of the cases, as discussed earlier (see Noonan and LEOPARD Syndromes above).63,64
Metabolic diseases also cause HCM phenocopy. Refsum disease, Pompe disease (glycogen storage disease type II), Danon disease, Niemann-Pick disease, Gaucher disease, hereditary hemochromatosis, and CD36 deficiency are examples of metabolic disorders that cause HCM phenocopy.186-189 Defective mitochondrial oxidative phosphorylation pathways also cause HCM phenocopy. Kearns-Sayre syndrome is a mitochondrial disease characterized by a triad of progressive external ophthalmoplegia, pigmentary retinopathy, and cardiac conduction defects and less frequently HCM phenocopy.117
In keeping with the diversity of cardiac phenotypes in HCM, expression levels of a variety of genes, in response to the mutant protein, are altered. Expression of genes encoding contractile sarcomeric proteins, cytoskeletal proteins, ion channels, intracellular signaling transducers, proteins maintaining the reduction–oxidation state, and those involved in transcriptional and translation machinery are changed.190,191 Expression levels of the markers of “secondary” cardiac hypertrophy, such as skeletal α-actin, isoforms of myosin light chain, and atrial natriuretic factor, are upregulated.190 Upregulation of markers of secondary cardiac hypertrophy suggest that hypertrophy in HCM is also a “secondary” phenotype. Accordingly, common pathways are involved in induction of cardiac hypertrophy in genetic and acquired forms. The diversity of molecular phenotype is in accord with the diversity of pathologic and clinical phenotypes in HCM that encompass not only hypertrophy and disarray, but also interstitial fibrosis and others.
Collective data from genotype–phenotype correlation studies indicate that mutations exhibit highly variable clinical, ECG, and echocardiographic manifestations and that no particular phenotype is mutation specific.192 Observational data show that cardiac hypertrophy accelerates during puberty and adolescence in patients with HCM.193 The finding suggests that growth factors contribute to expression of cardiac hypertrophy. Similarly, experimental and clinical studies suggest that cardiac hypertrophy, the clinical hallmark of HCM, is a compensatory phenotype and likely to be modulated by a large number of genetic and nongenetic factors.194 Overall, the final phenotype in HCM is determined not only by the causal mutations but also by the effects of modifier genes, environmental factors, epigenetic and epistatic factors, and posttranscriptional and posttranslational modifications of the proteins.
Causal genes and mutations are the primary determinant of expressivity of cardiac phenotype, including the severity of hypertrophy and the risk of SCD.121,122,149,150,195-199 Collectively, the data suggest gene- and mutation-specific effects. In general, mutations in MYH7 are associated with an early onset, extensive hypertrophy, and a high incidence of SCD, which are variable among different MYH7 mutations.195,196,198,200 MYH7 mutations are considered major prognosticators in HCM (Fig. 82–5). Topography of the mutations and their impact on β-MHC protein function are likely to be important determinants of the severity of cardiac hypertrophy as well as the risk of SCD. However, it is important to note that there is a significant degree of variability, which is partly independent of the causal mutations and partly reflects the effects of modifier genes. Given the relatively low frequency of each causal mutation, results of genotype–phenotype correlation studies have had limited utility. Consistent correlations have been observed for only a few mutations, such as R403Q and R719W, which are associated with a high incidence of SCD and severe hypertrophy (see Fig. 82–5).195,201 In contrast, G256Q and L908V are associated with a benign and Q930L with an intermediate prognosis.195,196
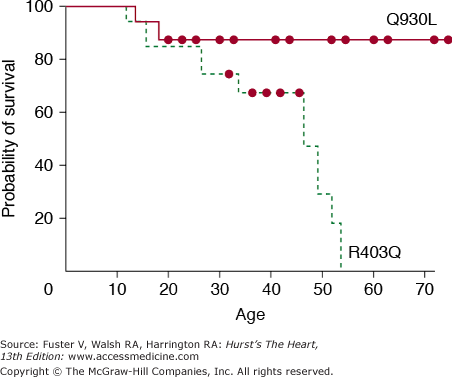
The phenotype in the majority of patients with MYBPC3 mutations is relatively mild. Commonly, the age of onset of clinical symptoms is late, the degree of cardiac hypertrophy is relatively mild, and the incidence of SCD is low.122,145,198 Accordingly, the penetrance, although age-dependent, is relatively low. Thus a normal physical examination, ECG, and echocardiogram have low negative predictive value in early life. The clinical phenotype often develops in the fifth or sixth decades of life. It may be unmasked by the presence of concomitant hypertension. Indeed, hypertensive HCM of the elderly could be a form of HCM caused by mutations in MYBPC3 and unmasked by hypertension.145 Nonetheless, there is a significant degree of variability in the phenotypic expression of HCM caused by MYBPC3, as “malignant” mutations, associated with severe hypertrophy and a high incidence of SCD, also have been described.122
The risk of SCD in HCM caused by mutations in MYH7 and MYBPC3 is partially reflective of the severity of hypertrophy.153 Mutations associated with mild hypertrophy generally carry a relatively benign prognosis, and those with severe hypertrophy indicate a high incidence of SCD. This is in contrast to HCM caused by mutations in TNNT2, which is characterized by mild cardiac hypertrophy, a high incidence of SCD, and extensive myocyte disarray.109,150 Inadequate genotype–phenotype correlation data are available regarding mutations in TNNI3, TPM1, TNNC1, TTN, ACTC, MYL3, and MYL2.
It is important to note that the results of genotype–phenotype correlation studies are subject to a large number of confounding factors, including the small size of the families, small number of families with identical mutations, low frequency of each mutation, phenotypic variability, homozygosity for the causal mutations or compound mutations, and the influence of modifier genes and environmental factors.158,202,203 Collective data indicate that mutations exhibit highly variable clinical, ECG, and echocardiographic manifestations, and no particular phenotype is mutation-specific.193
Expression of cardiac hypertrophy in HCM is modulated by interactions between the causal mutations, modifier genes, environmental factors, and epigenetic elements, as well as posttranscriptional and posttranslational modifications of proteins.194 Given the complexity of cardiac hypertrophy, a large number of genes are likely to modify phenotypic expression of HCM, each contributing a small fraction of the phenotype.
ACE I/D polymorphism, which is the most commonly studied polymorphism in HCM,162,165-171 accounts for approximately 3% to 5% of the total variability of left ventricular mass in genetically unrelated populations and for approximately 10% to 15% in members of the same family.174 Similarly, contributions of other potential modifier genes appears to be small.172-174
Evidence for the effect of environmental factors on cardiac phenotypes in HCM in humans, although expected, is circumstantial. Experimental data in animal models of HCM have not shown consistent effects.156,157,204 Because hypertrophy is considered a secondary phenotype, one could speculate that heavy physical exercise, particularly isometric exercise, could stimulate the development of severe hypertrophy in HCM. Although there is no direct evidence in humans, the common finding of HCM in young competitive athletes who succumb to SCD suggests that heavy physical exercise may worsen the cardiac phenotypes.
As the diversity of the causal mutations suggests, there is no single initial defect common to all mutations (Table 82–8). The diversity of the clinical phenotypes, such as hypertrophic, dilated, or restrictive cardiomyopathy arising from mutations in the same gene, further adds to the complexity of the pathogenesis. Topography of the causal mutation is likely to be important, as the initial defect is likely to be domain, but not protein, specific. Mutations located in a specific functional domain of a given protein are expected to confer similar initial defects, which may be organ-specific because of expression of the partner proteins. Given that each sarcomeric protein could have multiple functions, usually as a result of having multiple domains, mutations in the same protein could impart various initial defects.
| 1. Mechanical defect |
| Impaired actomyosin interaction |
| Impaired cardiac myocyte and myofibril contractile performance |
| 2. Biochemical defects |
| Impaired protein-protein binding |
| Impaired Ca2+ affinity of myofibrillar force generation |
| 3. Bioenergetics |
| Impaired myofibrillar adenosine triphosphatase activity |
| 4. Structural defects |
| Impaired sarcomere assembly |
| Impaired subcellular localization of sarcomeric proteins |
| Altered stoichiometry |
The causal mutation initiates a series of molecular events, which begins with alteration of the molecular structure and function of the protein (see Table 82–8). Because the majority of mutant sarcomeric proteins differ from the wild type only by a single amino acid (missense mutations), the mutant proteins incorporate into the sarcomere, albeit sometimes inefficiently.205 After incorporation, mutant sarcomeric proteins exert diverse functional defects, such as alterations in myofibrillar Ca2+ sensitivity and ATPase activity.206-210 Functional phenotypes lead to activation of secondary molecules, which are largely unknown but expected to include activating of many intracellular signaling pathways. The secondary molecular phenotypes mediate induction of the morphologic and histologic phenotypes. Accordingly, hypertrophy and fibrosis are considered secondary phenotypes because of activation of intermediary molecular phenotypes (Fig. 82–6).
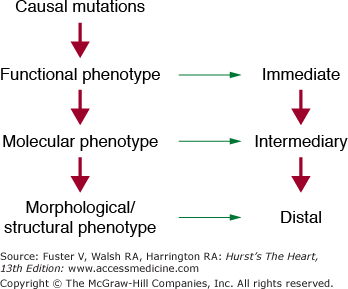
Many HCM mutations involve deletions or truncations that are considered null alleles because of the possible expression of unstable mRNA and proteins.143,211 Whether haploinsufficiency causes HCM by altering the stoichiometry of the sarcomeric proteins remains unknown. Myocyte dropout caused by apoptosis also has been implicated in animal models.212 However, the significance of myocyte apoptosis in the pathogenesis of human HCM remains to be established. Regardless of the initial primary defect, cardiac hypertrophy, the clinical hallmark of HCM, is considered a compensatory phenotype. Evidence suggesting the compensatory nature of cardiac hypertrophy includes upregulation of expression of molecular markers of secondary cardiac hypertrophy, such as atrial and brain natriuretic peptides,189,213,214 endothelin-1,215 transforming growth factor β1 (TGFβ1), and insulin-like growth factor 1 (IGF-1).216 The predominant involvement of the left ventricle and its frequent absence in the low-pressure right ventricle, despite equal expression of mutant sarcomeric protein in both, suggest contribution of the environment to the development of hypertrophy. Furthermore, variation in hypertrophic response because of the genetic background, its absence early on in life, and its attenuation through pharmacologic interventions, at least in animal models, supports the secondary nature of hypertrophy.217,218 The primary impetus for hypertrophy is not well defined. It is likely to involve activation of myofibrillar signaling pathways in response to increased cell mechanical stress, altered moiety of the mutant proteins for the signaling molecules, and/or altered Ca2+ homeostasis.
Current pharmacologic interventions in HCM are empiric (class IIa).82 None of the current pharmacologic agents have been shown to induce regression of hypertrophy, fibrosis, and disarray, which are associated with increased mortality and morbidity (class IIb).106,107 Currently, there is no suitable method to correct the underlying genetic defect. Therefore, the emphasis has been on prevention, reversal, and attenuation of the phenotype through pharmacologic interventions aimed at blockade of intermediary molecular phenotypes. Recent studies have shown potential clinical usefulness of angiotensin II receptor blockers, beta-hydroxy-beta-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors, and antioxidants in prevention, attenuation, and reversal of cardiac phenotypes in animal models of HCM (class IIb).217-220 For example, blockade of angiotensin II receptor 1 in cardiac troponin T-Q92 transgenic mice reduced interstitial collagen volume, expression levels of collagen α1 (I) mRNA, and TGFβ1 protein to normal levels.219 Treatment with HMG-CoA reductase inhibitors also has been shown to prevent and attenuate cardiac phenotype in a transgenic rabbit model of human HCM.217,218 Treatment with simvastatin, a pleiotropic HMG-CoA reductase inhibitor, reduced left ventricular mass, wall thickness, and collagen volume fraction in the β-MHC-Q403 transgenic rabbit model of human HCM.217 In addition, indices of left ventricular filling pressure were improved significantly. Similarly, administration of atorvastatin prevented the development of cardiac hypertrophy in the same transgenic rabbit model.218 Treatment with antioxidant N–acetylcysteine (NAC) reversed interstitial fibrosis in a mouse model of HCM.220 Likewise, treatment with NAC resolved cardiac hypertrophy and fibrosis in a transgenic rabbit model of HCM and reduced vulnerability to cardiac arrhythmias.221 Calcineurin inhibitors, although effective in attenuation of experimental cardiac hypertrophy,222 were found to worsen cardiac phenotype in an α-MHC mouse model of HCM.206 Pretreatment with diltiazem, an L-type Ca2+ channel blocker, prevented the exaggerated cardiac hypertrophic response to calcineurin inhibitors in the α-MHC mouse model.206 Collectively, the results in genetically engineered animal models raise the possibility of pharmacologic interventions to prevent, attenuate, and reverse evolving cardiac phenotype in HCM. Clinical trials in humans are needed to determine potential salutary effects of HMG-CoA reductase inhibitors, angiotensin II blockers, and N-acetylcysteine in human patients with HCM.
DCM is a primary disease of the myocardium that manifests by dilatation of the left ventricular along with a gradual decline in contractility. The diagnosis is based on a left ventricular ejection fraction of less than 0.45 and a left ventricular end-diastolic diameter of more than 2.7 cm/m2. It has a prevalence of 40 cases per 100,000 individuals and an incidence of 5 to 8 cases per 100,000 persons.223 Patients with DCM are often asymptomatic in the early stages but gradually develop symptoms and signs of heart failure, syncope, cardiac arrhythmias, and SCD. Thus a normal history and physical examination in a subject at risk, particularly in the early decades of life, does not exclude DCM. A significant number of affected relatives of patients with DCM are asymptomatic and are diagnosed for the first time on additional testing (such as an echocardiogram).224 A family history of DCM is present in approximately half of all index cases with idiopathic DCM.224,225 In the remainder, DCM is considered sporadic. Familial DCM is commonly inherited as an autosomal-dominant disease,224 which clinically manifests during the third and fourth decades of life.
An X-linked DCM is suspected when only male members of a family exhibit symptoms and signs of DCM and there is no male-to-male transmission. Three common forms of X-linked DCM have been identified, including Duchenne and Becker muscular dystrophies, Emery-Dreifuss syndrome, and Barth syndrome. DCM also occurs in multiorgan disorders, such as mitochondrial DNA mutations, triplet repeat syndromes, and metabolic disorders.
DCM is an extremely heterogeneous disease, as indicated by the heterogeneity of the mapped loci and genes for familial DCM (Table 82–9). The predominant mode of inheritance is autosomal dominant; however, autosomal-recessive and X-linked DCM also occur. Several causal genes for autosomal-dominant DCM have been identified.220 Several causal genes encode sarcomeric proteins, which are also known to cause HCM.221 Thus, despite the contrasting phenotypes of HCM and DCM, mutations in sarcomeric genes can cause either of the phenotypes. Because many of the known causal genes for DCM involve the myocyte cytoskeleton, DCM has been considered to be a disease of cytoskeletal proteins. However, mutations in proteins other than the cytoskeletal proteins also can cause DCM.
| Gene | Symbol | Locus | Inheritance | Mutations/Frequency/Context |
|---|---|---|---|---|
| Sarcomeric/cytoskeletal | ||||
| Cardiac α-actin | ACTC | 15q11-14 | Autosomal dominant | Missense/uncommon; also causes HCM |
| β-Myosin heavy chain | MYH7 | 14q11-13 | Autosomal dominant | Missense/∼5%; also causes HCM |
| Cardiac troponin T | TNNT2 | 1q32 | Autosomal dominant | Missense/uncommon; also causes HCM |
| α-Tropomyosin | TPM1 | 15q22.1 | Autosomal dominant | Missense/rare; also causes HCM |
| Cypher/ZASP (LIM domain binding 3) | LDB3 | 10q22.3-q23.2 | Sporadic and familial | |
| Titin | TTN | 2q24.1 | Autosomal dominant | Missense/uncommon; also causes HCM |
| Telethonin (T-cap) | TCAP | 17q12 | ||
| Cytoskeletal | ||||
| α-Sarcoglycan | SGCA | 17q21 | Autosomal dominant | Limb–girdle muscular dystrophy |
| β-Sarcoglycan | SGCB | 4q12 | Autosomal dominant | |
| α-Sarcoglycan | SGCD | 5q33-34 | Autosomal dominant | |
| Autosomal recessive | ||||
| Dystrophin | DMD | Xp21 | X-linked | Muscular dystrophy |
| Muscle LIM protein | MLP (CSRP3) | 11q15.1 | Autosomal dominant | Rare, founder effect in families described |
| Ankyrin repeat domain 1 | ANKRD1 | 10q23.31 | Sporadic | ∼1% |
| Intermediary filaments | ||||
| Desmin | DES | 2q35 | Autosomal dominant | Also causes RCM and desminopathies |
| α-B-Crystallin | CRYAB | 11q35 | Desminopathy | |
| Nuclear proteins | ||||
| Lamin A/C | LMNA | 1q21.2 | Autosomal dominant | DCM, conduction defect, muscular dystrophy, lipodystrophy, insulin resistance |
| Emerin | EMD | Xq28 | X-linked | |
| Vinculin | VCL | 10q22.1-q23 | Sporadic | Metavinculin isoform |
| Cell junction molecules | ||||
| Desmoplakin | DSP | 6p23-25 | Autosomal recessive | Also causes ARVC |
| Unknown | ||||
| Taffazin (G4.5) | TAZ | Xq28 | X-linked | Ventricular noncompaction |
| 1q32 | ||||
| 2q14-22 | ||||
| 2q31 | ||||
| 3p22-25 | ||||
| 6q23-24 | ||||
| 9q13-22 | ||||
| 10q21-23 | Autosomal dominant | |||
| 7p12.1-7q21 | ||||
The gene encoding cardiac α-actin (ACTC) was the first causal gene identified for autosomal-dominant DCM.228 Subsequently, mutations in genes encoding additional components of the sarcomere, namely MYH7, TNNT2, TTN, and TCAP, were found in patients with DCM.225,226,229 Because mutations in ACTC,MYH7, and TNNT2 are also known to cause HCM, these findings point to the commonality of the genetic basis of DCM and HCM. The diversity of the phenotype may reflect the topography of the causal mutations on the protein, as well as the genetic background of the individuals.
Mutations in cytoskeletal proteins—namely, delta sarcoglycan,230 beta sarcoglycan,231 metavinculin,232 and dystrophin233—are also important causes of DCM. Mutations in alpha sarcoglycan (adhalin) cause an autosomal-recessive form of DCM that occurs in conjunction with limb-girdle muscular dystrophy.231 Recently, a mutation in muscle LIM protein (MLP) was identified in several related families with DCM.229 MLP interacts with telethonin, a titin-interacting protein, and colocalizes with it to the Z disk. Mutations in another Z disk protein named ZASP or Cypher or LIM domain binding protein 3 (LDB3) also have been identified in patients with DCM.234,235 These findings, together with the results of studies in the LIM-deficient mouse model, implicate the Z disk is a mechanosensor for cardiac myocytes. Furthermore, rare mutations in ABCC9, which encodes the regulatory SUR2A subunit of the cardiac K(ATP) channel, have been identified in patients with DCM.236 Mutant SUR2A proteins show aberrant redistribution of conformations in the intrinsic adenosine triphosphate (ATP) hydrolytic cycle and induce abnormal K(ATP) channel phenotypes.236
An intriguing causal gene for familial DCM is the lamin A/C gene,237 which encodes a nuclear envelope protein. The observed phenotype resulting from mutations in the rod domain of lamin A/C is progressive conduction disease, atrial arrhythmias, heart failure, and SCD. Moreover, mutations in the intermediary filament desmin and its associated protein alphaB-crystallin have been identified in patients with DCM.238,239 Often such mutations lead to a phenotype of cardiac and skeletal myopathy that is referred to as desmin-related myopathy.239 Collectively, these findings suggest that mutations affecting the integrity of the sarcomeric, cytoskeleton, and Z disk proteins are the main causes of DCM.
The phenotype is characterized by progressive degeneration of muscle function. It commonly manifests itself as mild but progressive skeletal myopathy, early contractures, and cardiomyopathy. The incidence of Duchenne/Becker muscular dystrophy is 1 in 3500 newborn males.240 Duchenne muscular dystrophy is a severe form and Becker muscular dystrophy a milder form of the disease. The disease commonly manifests itself during the first or second decades of life in male patients. Female family members are commonly spared. However, they may exhibit a mild phenotype, typically late in life. Cardiac involvement includes progressive atrioventricular block, arrhythmia, loss of P-wave amplitude on the ECG, atrial standstill, DCM, akinesis/dyskinesis of the posterobasal wall of the left ventricle, and SCD. Often, DCM is the primary feature of Duchenne and Becker muscular dystrophies. Approximately 90% of patients will eventually develop DCM.241 Death often occurs by the third decade of life.
The gene responsible for Duchenne and Becker muscular dystrophies is dystrophin, located on Xp21, which encodes a large cytoskeletal protein.242 A variety of point, deletion, and insertion mutations or gene rearrangements in dystrophin have been described.233,241 Approximately two-thirds of the mutations are either deletions or duplications. DCM can also occur in the absence of skeletal myopathy. Mutations leading to a frameshift induce a severe form, whereas missense mutations often lead to a mild form of the disease. Mutations in the 5′ region of the dystrophin gene can cause DCM without skeletal involvement.243
Emery-Dreifuss muscular dystrophy is an X-linked degenerative disorder characterized by mild but progressive skeletal and cardiac myopathy.244,245 Clinical features include muscle weakness and atrophy, flexion deformities of the elbows, and mild pectus excavatum. Cardiac phenotypes include cardiomyopathy, arrhythmia, SCD, conduction defects, loss of P-wave amplitude on the ECG, and atrial standstill.246
The causal gene is EMD, which encodes emerin. Emerin is located along the nuclear rim of many cell types. It is a member of the nuclear lamina-associated protein family.247
DCM is also a major phenotypic component of the Barth syndrome, another X-linked disorder. The characteristic phenotype of Barth syndrome includes skeletal and cardiac myopathy, neutropenia, and abnormal mitochondria. Barth syndrome is caused by point, deletion, and splice-junction mutations in the tafazzin (TAZ) or G4.5 gene, located on Xq28.248
The phenotype of DCM is determined not only by the causal mutations, but also by the modifier genes and the environmental factors. Genetic studies to identify the modifier genes for DCM are largely restricted to SNP-association studies and have limitations similar to those described for HCM. Several potential candidates, including ACE, have been identified. None have been established to modify cardiac phenotypes in DCM. A modifier locus in a calsequestrin mouse model of DCM has been mapped. 249
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


