18
Cardiomyopathies
 Cardiomyopathy
Cardiomyopathy
Cardiomyopathies are a heterogeneous group of diseases of the myocardium associated with mechanical and/or electrical dysfunction that usually (but not invariably) exhibit inappropriate ventricular hypertrophy or dilatation and are due to a variety of causes that frequently are genetic. Cardiomyopathies either are confined to the heart or are part of generalized systemic disorders, often leading to cardiovascular death or progressive heart failure–related disability. 1
This new classification divided cardiomyopathies into two major groups—primary and secondary—based on the predominant organ involvement. It retained the common clinical cardiomyopathies synonymous with worsening myocardial performance due to diastolic or systolic dysfunction, but for the first time included diseases with electrical abnormalities that cause life-threatening arrhythmias (differentiated by their specific molecular nature). Primary cardiomyopathies include those conditions with only or mostly myocardial involvement (genetic, mixed, acquired) ( Fig. 18-1). This places a traditional cardiomyopathy like hypertrophic cardiomyopathy (HCM) with disorders that cause tachyarrhythmias because of genomic changes that encode for ion channel dysfunction. 1 Secondary cardiomyopathies are defined as the result of any disease process that includes the heart but is not limited to the heart. These were previously referred to as specific cardiomyopathies or specific heart muscle diseases. Box 18-1 lists some of the major disease processes that may be associated with cardiomyopathy but are not considered “primary” cardiomyopathies under the new classification. Also excluded are cardiomyopathies due to myocardial conditions such as valvular, congenital heart, and atherosclerotic disease. The common use of “ischemic cardiomyopathy” is excluded.
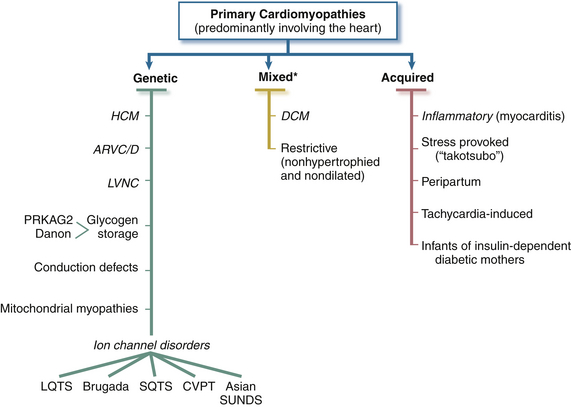
Figure 18-1 Primary cardiomyopathies in which clinically relevant disease processes solely or predominantly involve myocardium. Conditions have been segregated according to their genetic or nongenetic etiologies.∗ Predominantly nongenetic; familial disease with a genetic origin has been reported in a minority of cases. (From Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association scientific statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807-1816.)
Enormous work has been done identifying genetic causes for cardiomyopathies. Genetic testing is advancing rapidly to identify disease-causing mutations in family members at risk but asymptomatic. 2 The primary cardiomyopathies have a very complex genetic profile, with mutations that affect clinical expression of the same disease. The result is heightened clinical surveillance and possibly earlier intervention and prevention of the sequelae of cardiomyopathies. At this time, genetic testing is not 100% sensitive, so it is usually performed once the disease has been diagnosed and confirmed clinically. Genetic screening’s greatest use is to identify carriers within a family. This may allow family members that carry the same genetic mutation to be followed, since they may have a reduced penetrance that leads to a lesser form of the disease or even asymptomatic expression. 3
Most inherited cardiomyopathies demonstrate a mendelian autosomal dominant inheritance. It has been accepted that HCM is largely a genetic defect of the contractile proteins. In contrast, the genetic trail of dilated cardiomyopathy (DCM) is really only solid as it pertains to familial DCM. The more common sporadic DCM has not been found to have a genetic basis. Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) is primarily related to genetic mutations that encode proteins of the desmosome. Desmosomes, cell-cell adhesion organelles, are especially abundant in heart tissue. 4 A genetic basis for restrictive cardiomyopathy (RCM) has not been identified, but a familial form exists and is caused by mutations in the troponin I gene. 5 The genetic basis for left ventricular noncompaction (LVNC) has not been found (and the diagnostic criteria for LVNC continue to be debated), 6 but it is frequently familial, with at least 25% of asymptomatic relatives having a range of echocardiographic abnormalities. 5 More extensive information about the genetics of primary cardiomyopathies is available.7–10
 Dilated Cardiomyopathy
Dilated Cardiomyopathy
Formerly referred to as congestive cardiomyopathy or idiopathic cardiomyopathy, DCM is by far the most common of the four major cardiomyopathies in adults (60%), with a prevalence of 1:2500 individuals. 1 It is the third leading cause of heart failure overall, with nearly 550,000 individuals diagnosed each year. Not surprisingly, it is the most common indication for cardiac transplantation, because survival for adults at 1 and 5 years after diagnosis is 76% and 35%, respectively. 11 Since DCM was classified as a primary cardiomyopathy in 2006, the term idiopathic has rarely been applied, because the heart is the main organ involved. The disease process is of mixed etiology; both genetic and acquired cases have been described. 1 It is now appreciated that there is a genetic basis for DCM when all the more common causes have been excluded. More recently, the familial prevalence rate of DCM has been shown to range from 20% to 50%. 8 More than 20 genes have been identified as causes of DCM. Most genetic inheritance is autosomal dominant. The familial form of DCM shows age-dependent penetrance, so that even a normal echocardiogram does not exclude onset later in life. 10 Incomplete penetrance may account for differences in disease severity and progression in familial DCM, despite identical mutations. 12 Interestingly, 30% of patients diagnosed with DCM are asymptomatic. 13 If medical treatment is begun immediately, there is extended quality of life and survival for those individuals. Improved survival supports the vast potential benefits of genetic research in cardiomyopathies.
Numerous causes of acquired DCM exist that include a variety of viral, bacterial, and parasitic infections; autoimmune disorders; neuromuscular diseases (Duchenne muscular dystrophy); exposure to toxic agents, including chemotherapeutic drugs, ethanol, mercury, and lead; as well as certain dietary deficiencies. In North America, myocarditis is the major cause of DCM, usually secondary to a viral illness. 12
DCM is characterized morphologically by right and left ventricular cavity enlargement, with hypertrophied muscle fibers without an appropriate increase in the ventricular septal or free wall thickness, giving an almost spherical shape to the heart. The heart is often two to three times larger than normal. 12 The valve leaflets may be normal, but dilation of the heart may cause a regurgitant lesion secondary to displacement of the papillary muscles and tethering of the chordae tendineae. Histologic changes are nonspecific and not associated with positive immunohistochemical, ultrastructural, or microbiological tests. Microscopically, instead of large losses of myocardium, there is patchy and diffuse loss of tissue, with interstitial fibrosis and scarring uncharacteristic of ischemic myocardium. 12
With DCM, there is more impairment of systolic function even though diastolic function is affected. As contractile function diminishes, stroke volume is initially maintained by augmentation of end-diastolic volume. Despite a severely decreased ejection fraction (EF), stroke volume may be almost normal. Eventually, increased wall stress due to marked left ventricular (LV) dilation and normal or thin LV wall thickness occurs. 14 The echocardiographic diagnostic criteria for DCM are a dilated LV ( Fig. 18-2) together with reduced systolic function and normal or thin walls ( Fig. 18-3). However, for greater diagnostic accuracy, the patient should not have abnormal LV loading conditions (e.g., severe valvular heart disease, significant CAD). 15 Some experts have suggested specific diagnostic criteria for measurements of LV size and systolic function that include an LVEF less than 45%, a fractional shortening less than 25%, and an LV end-diastolic diameter greater than 112%. 15 Criteria should be corrected for age, gender, and body surface area ( Box 18-2).
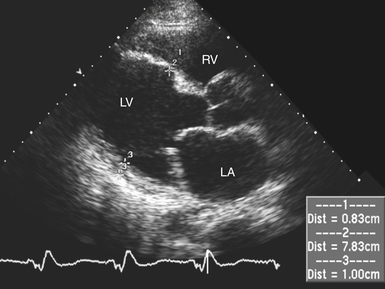
Figure 18-2 Transthoracic parasternal long-axis image of patient with dilated cardiomyopathy. In this diastolic frame, left ventricle is noted to be severely enlarged with normal wall thickness. LA, Left atrium; LV, left ventricle; RV, right ventricle.
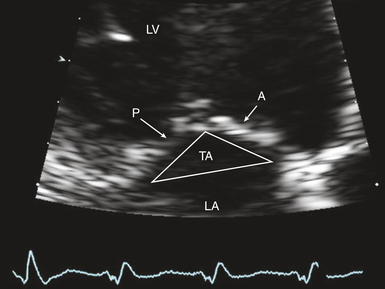
Figure 18-3 Zoomed transthoracic apical long-axis view of mitral valve showing anterior (A) and posterior (P) leaflets. Note significant valve tenting; tenting area (TA) is outlined. LA, Left atrium; LV, left ventricle.
Increasing left atrial (LA) size may indicate worsening diastolic dysfunction in these patients contributing to functional mitral regurgitation (MR). 16 Dilation combined with valvular regurgitation compromises the metabolic capabilities of heart muscle and produces overt circulatory failure. Compensatory mechanisms may allow symptoms of myocardial dysfunction to go unnoticed for an extended period of time. However, the onset of MR signals a poor prognosis because ventricular function progressively worsens without intervention. The importance of corresponding neurohumoral influences (e.g., renin-angiotensin system) in this pathologic process has only recently been appreciated as a major factor in the appearance of common signs and symptoms of congestive heart failure (CHF) and development of therapeutic options ( Fig. 18-4).17,18
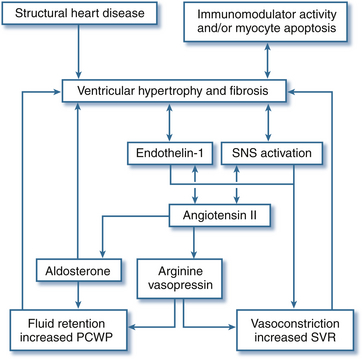
Figure 18-4 Impact of pathophysiologic mediators on hemodynamics in patients with heart failure. PCWP, Pulmonary capillary wedge pressure; SNS, sympathetic nervous system; SVR, systemic vascular resistance. (From McBride BF, White CM. Acute decompensated heart failure: a contemporary approach to pharmacotherapeutic management. Pharmacotherapy. 2003;23:1002.)
DCM usually presents in the fourth and fifth decades of life. 12 The clinical picture typically includes signs and symptoms of CHF often corresponding to months of fatigue, weakness, and reduced exercise tolerance prior to diagnosis. 15 One third of individuals complain of chest pain. 14 However, the first indication of DCM may be a stroke, arrhythmia, or even sudden death. Increasingly, individuals presenting for routine medical screening are found to have cardiomegaly on a routine chest roentgenogram. Symptoms may appear insidiously over a period of years or evolve rapidly after an unrelated illness. Physical signs of DCM depend on the disease’s progression but may include pulsus alternans, jugular venous distention, murmurs of atrioventricular valvular regurgitation, tachycardia, gallop heart sound, rales, cool extremities, and leftward displacement of the apex with palpation of the precordium.
A chest roentgenogram demonstrates variable degrees of cardiomegaly and pulmonary venous congestion ( Fig. 18-5, A). An electrocardiogram (ECG) may be surprisingly normal or depict low QRS voltage, abnormal axis, nonspecific ST-segment abnormalities, LV hypertrophy, conduction defects, and evidence of atrial enlargement. Atrial fibrillation is common, and about a fourth of patients have nonsustained ventricular tachycardia (VT). 15 B-type natriuretic peptide (BNP) levels also increase dramatically with ventricular wall stress and are predictive of mortality in acute decompensated CHF. 19 Coronary catheterization would reveal marked LV dilation ( Fig. 18-5, B) and typically normal coronary vessels, which has therapeutic and prognostic implications. An endomyocardial biopsy is rarely valuable or indicated to identify the etiology of DCM but may be useful to rule out other pathologies with presentations similar to DCM. 12
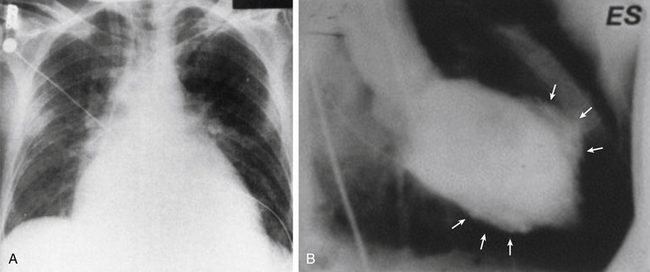
Figure 18-5 A, Chest radiography showing marked cardiomegaly in patient with AIDS who developed dilated cardiomyopathy following treatment for Pneumococcus carinii pneumonia. B, Left ventriculogram of different patient with sarcoidosis and dilated cardiomyopathy at end-systole, demonstrating uneven wall motion abnormalities with discrete dyskinetic or akinetic regions (arrows). There is marked dilation even in end-systole. ES, End-systole. (A from Corboy JR, Fink L, Miller WT. Congestive cardiomyopathy in association with AIDS. Radiology. 1987;165:139-141. B from Yazaki Y, Isobe M, Hiramitsu S, et al. Comparison of clinical features and prognosis of cardiac sarcoidosis and idiopathic dilated cardiomyopathy. Am J Cardiol. 1998;82:537-540.)
The approach to diagnosing DCM with echocardiography begins by demonstrating LV chamber enlargement and systolic dysfunction in the setting of normal or reduced wall thickness (Videos 18-1 and 18-2  ). Indeed, all markers of systolic function (EF, fractional shortening, stroke volume, cardiac output [CO]) are uniformly decreased. 20 Specific diagnostic echocardiographic criteria have been noted (see Box 18-2).
). Indeed, all markers of systolic function (EF, fractional shortening, stroke volume, cardiac output [CO]) are uniformly decreased. 20 Specific diagnostic echocardiographic criteria have been noted (see Box 18-2).
With progressive dilation, LV geometry changes and the LV assumes a more spherical rather than ellipsoid shape. Alterations in mitral valve geometry accompany ventricular remodeling. Geometric abnormalities resulting from LV remodeling can be quantitated in patients with DCM. In particular, the sphericity index allows echocardiographers to quantitate the progression of LV shape from ellipsoid to spherical that characterizes DCM. More than one method has been reported for calculating the sphericity index, potentially leading to confusion during report interpretation. One method calculates the sphericity index as the calculated LV volume divided by the volume of a sphere whose diameter is the major-axis (long-axis) linear measurement of the LV. 21 Others use simple linear measurements rather than volumes. Matsumoto et al. calculated sphericity index as the ratio of the LV major axis (long axis) to the LV minor axis (short axis) as determined from a four-chamber echocardiography view. 22 These two methods yield different results, with the former producing a value less than 1 and the latter generating a value greater than 1. For instance, Matsumoto used simple linear measurements and noted that the sphericity index in patients with DCM averaged 1.5 ± 0.2 while the index in normal controls averaged 1.9 ± 0.3. 22 Sadeghpour et al. 23 showed that increases in sphericity parallel worsening of functional MR.
Functional MR is a common finding in patients with DCM. Despite structurally normal leaflets, remodeling of the LV leads to changes in the mitral apparatus. The mitral annulus dilates and the coaptation point of the leaflets occurs closer to the apex, leading to the appearance of valve tenting (see Fig. 18-3). Both the tenting area and tenting distance can be measured ( Fig. 18-6). The degree of MV tenting correlates well with functional MR severity. Sadeghpour et al. 23 noted that tenting distance correlated more closely with functional MR severity than annular dilatation or sphericity index. In particular, these investigators found that in patients with mild functional MR, the tenting distance (distance from the mitral leaflet coaptation point to the plane of the mitral annulus) averaged 1.1 ± 0.25 cm. However, the tenting distance in patients with severe functional MR averaged 1.56 ± 0.25 cm (P < 0.001).
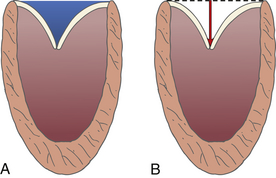
Figure 18-6 A, Mitral valve tenting area blue shaded area. B, Tenting distance, or distance from plane of mitral annulus to leaflet coaptation point (arrow). (Used with permission of Mayo Foundation for Medical Education and Research. All rights reserved.)
A thorough echocardiographic examination in a patient with DCM should include assessment of right ventricular (RV) size and systolic function. Variable RV involvement in the disease process characterizes patients with DCM. RV dysfunction (when present) tends to be associated with worse LV systolic function 15 and is associated with a greater degree of heart failure symptoms and worse clinical outcome. 24 Similarly, assessment of diastolic function may be helpful. Faris et al. noted that patients with DCM who also have a restrictive diastolic filling pattern have a mortality rate three times higher than DCM patients without a restrictive filling pattern. 25 The usual methods of diastolic assessment include Doppler flow patterns of mitral inflow, pulmonary veins, and tissue Doppler interrogation of the mitral annulus (see Chapter 12).
Intracardiac thrombus formation is possible in the setting of reduced LV systolic function due to intracavitary stasis. A comprehensive echocardiographic examination in patients with DCM should include a search for intracardiac thrombus, particularly in the LV apex and LA appendage. However, the risk of thromboembolic complications is relatively low (1%-3%/yr) even with very poor EF and echocardiographic evidence of intracardiac thrombi. 26 The rates are so low that it is difficult to determine a difference in outcome with anticoagulation. Patients with DCM may be on anticoagulation if they have experienced a prior embolic event or paroxysmal or persistent atrial fibrillation, 27 but the benefit remains unclear.
Color Doppler imaging is also useful in assessing the presence or absence of valvular regurgitation. Pulsed wave and continuous wave Doppler (PWD, CWD) are used to quantify CO and evaluate filling pressures and pulmonary artery pressures. Well-compensated patients with DCM may have only mild impairment of diastolic function. As the disease progresses and patients become less well compensated, the LV diastolic filling pattern changes to that of restricted filling. While systolic function may not change in these patients, the increased filling pressures associated with restrictive LV filling will often worsen their CHF symptomatology.
Compared to ischemic CHF, patients with nonischemic DCM show greater improvement in symptoms, LV function, and remodeling with contemporary therapy than in the past. 28 Guidelines are available for clinical care of patients with DCM along the entire spectrum of the disease, presymptomatic to end stage. 26 Treatment revolves around management of symptoms and progression of DCM, while other measures are designed to prevent complications such as pulmonary thromboembolism and arrhythmias. The mainstay of therapy for DCM is an ACE inhibitor or angiotensin II receptor blocker, diuretic, and a β-blocker. Recent trials have supported selective use of spironolactone in DCM owing to its association with improved survival, but renal function must be carefully followed. 26 Afterload-reducing agents such as selective phosphodiesterase-3 inhibitors (e.g., milrinone) may improve quality of life but do not affect mortality so are rarely administered chronically in DCM. Digoxin is not considered a first-line drug for CHF and DCM but may play a role in a few selected patients, 26 since it has been clinically beneficial in two large adult trials. 29
Use of β-blockers in DCM has not only provided symptomatic improvement but substantial reductions in sudden and progressive death in New York Heart Association (NYHA) class II and III heart failure. 30 This is especially significant because almost 50% of deaths are sudden. 31 Experience with β-blockers suggests that if taken long term, symptoms improve and there is improvement in both clinical status and overall sense of well-being. Like the angiotensin-converting enzyme (ACE) inhibitors, β-blockers reduce risk of death and the combined risk of death and hospitalizations. 26
High-grade ventricular arrhythmias are common with DCM. Some 12% of all patients with DCM die suddenly, 32 but overall prediction of sudden death in an individual with DCM is poor. 31 Electrophysiologic (EP) testing has a poor negative predictive value that limits its usefulness. The best predictor of sudden death remains the degree of LV dysfunction. Patients who have sustained VT or out-of-hospital ventricular fibrillation are at increased risk for sudden death, but more than 70% of patients with DCM have nonsustained VT during ambulatory monitoring. 33 Prophylactic administration of antiarrhythmic drugs to suppress premature ventricular depolarizations and nonsustained ventricular arrhythmias has not been shown to improve survival. 26 Consequently, antiarrhythmic agents should not be considered part of routine treatment in patients with DCM, even with frequent premature ventricular depolarizations or asymptomatic nonsustained VT. Furthermore, most antiarrhythmic medications have a negative inotropic effect. Amiodarone is the safest agent and most effective when antiarrhythmic therapy is necessary to prevent recurrent atrial fibrillation or symptomatic ventricular arrhythmias. 26
The role of the implantable cardioverter-defibrillator (ICD) is complex in these patients in terms of balancing the benefits with the risks. There is evidence that with previous cardiac arrest or sustained VT, more benefit was gained from an ICD. 34 More recently, Kadish et al. 35 enrolled 458 patients with nonischemic DCM, LVEF less than 36%, and premature ventricular complexes or nonsustained VT. A total of 229 patients were randomly assigned to receive standard medical therapy, and 229 to receive standard medical therapy plus a single-chamber ICD. There were 17 sudden deaths from arrhythmia: 3 in the ICD group and 14 in the standard therapy group (P = 0.006). In this population of patients, the ICD significantly reduced the risk of sudden death, and there was a nonsignificant reduction in risk of death from any cause (P = 0.08). Complications associated with ICD insertion (e.g., bleeding, infection) can be serious. The study could not say that ICDs should be routinely placed in all patients with DCM, but should be considered on a case-by-case basis.
Patients who are resistant to pharmacologic therapy for CHF with DCM have received left ventricular assist devices (LVAD), cardiac surgery (mitral valve repair or replacement), and transplantation in recent years. Transplantation is still the most viable surgical approach to end-stage DCM and can substantially prolong lives, 36 but limited organ availability and drug-related morbidity looks to future improvements in assist devices for increased survival. Mechanical devices like LVADs have become implantable, so discharge from the hospital is possible. LVADs are currently used primarily as “bridges” to transplantation, but they have also been considered as “destination” therapy in some patients who are not candidates for transplantation. Completion of a study looking at destination therapy with LVADs compared to medical management in patients with DCM who were ineligible for transplant showed a 2-year survival of 23% with an LVAD, compared to 8% of patients who received only medical therapy. 37 Although efficacy was demonstrated, there are a number of serious complications such as bleeding, stroke, sepsis, and device failure. Occasionally the mechanical device can be removed several months after placement in the patient if the resting ventricle has recovered enough for explantation.
Finally, cardiac surgery is not too dangerous when necessary for patients even with severely depressed EF and DCM. Sometimes if MR develops in an individual with DCM, mitral valve repair or replacement is safe to perform and has been shown to improve NYHA classification and survival. 38
 Hypertrophic Cardiomyopathy
Hypertrophic Cardiomyopathy
HCM is the most common genetic cardiac disease, with marked heterogeneity in clinical expression, pathophysiology, and prognosis. The overall prevalence for adults in the general population is 0.2%, 39 affecting men and women equally. It is the most common cause of sudden death in athletes, yet most HCM patient have a normal life expectancy without disability or the need for major therapeutic interventions. 40
HCM is a disease of the cardiac sarcomere. This is a primary myocardial disease with myocyte disarray, diastolic dysfunction, and asymmetric LV hypertrophy ( Fig. 18-7, A). The extent of sarcomeric disarray distinguishes HCM from other conditions, but this pattern is also seen in pressure-overloaded ventricles. 41 The hypertrophied muscle is composed of cells with bizarre shapes and multiple intercellular connections arranged in a chaotic “whorling” pattern characteristic of HCM ( Fig. 18-7, B).39,42 Increased connective tissue combined with markedly disorganized and hypertrophied myocytes contribute to the diastolic abnormalities present in all HCM patients. It manifests as a stiffer ventricular chamber, impaired and prolonged relaxation of the ventricular muscle, severely impaired filling, and an unstable electrophysiologic substrate that causes complex arrhythmias and sudden death. 39 Doppler imaging techniques may be utilized to evaluate diastolic function in patients with HCM, but many of the parameters typically used to grade diastolic function are less reliable in HCM than in other disease states. In particular, E-wave deceleration time and transmitral E/A ratios tend to correlate poorly with LV end-diastolic pressure in HCM. 43 Pulmonary vein velocities also appear less reliable in HCM and in other cardiomyopathies. The E/e′ (tissue Doppler imaging–derived e′) appears more reliable in estimating left-sided filling pressures, 44 but in one study, 25% of patients with an E/e′ ratio above 15 still had LA pressures less than 15 mmHg. 45 It is worth noting that in patients with normal LA size (LA volume index ≤28 cm3/m2) LV filling pressures are likely to be normal. 46
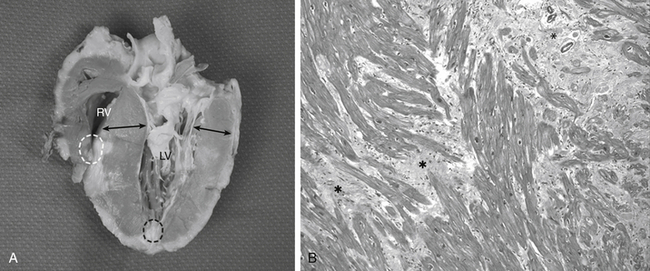
Figure 18-7 A, Hypertrophic cardiomyopathy (HCM) from saggital section of explanted native heart. Arrows indicate asymmetric myocardial thickening. Ventricular cavity is small. Thickening is predominately in subaortic region but also occurs in midventricular and apical regions as well as lateral and posterior wall. Apical aneurysms in left (black dashed circle) and right (white dashed circle) ventricles are observed. Note narrowing of left ventricular outflow tract caused by thickened ventricular septum and anterior leaflet of mitral valve. B, Photomicrograph of septal myocardium in HCM. Note pronounced disarray and hypertrophy of individual muscle fibers, resulting in a “whorled” appearance. Normal myocytes run in straight parallel bundles, but these often are identified as running at oblique angles. Interstitial fibrosis is found in many areas. In this figure, with increased magnification, myofibril disarray is evident in many of myocytes. Variation in size of nuclei is also evident. ∗Areas of significant interstitial myocardial fibrosis. (Movat pentachrome stain ×6400.) (From Soor GS, Luk A, Ahn E, et al. Hypertrophic cardiomyopathy: current understanding and treatment objectives. Clin Pathol. 2009;62:226-235.)
Besides diastolic dysfunction, the other major abnormality and fundamental characteristic of HCM is unexplained LV hypertrophy in a nondilated ventricular chamber in the absence of another cardiac or systemic disease capable of generating this degree of muscle hypertrophy. 1 This nonuniform asymmetric hypertrophy typically occurs in the basal anterior ventricular septum and anterior free wall, with a disproportionate increase in ventricular wall thickness relative to the posterior free wall, as shown in the gross specimen in Figure 18-7, A. The extent and pattern of hypertrophy varies greatly. The LV wall thickness is the most extensive of all cardiac conditions, 39 yet heart size may be deceptive in that it may vary from normal to more than 100% enlarged. Increases in wall thickness, not chamber enlargement, are responsible for the increase in ventricular mass.
Two-dimensional (2D) and Doppler echocardiography easily identify LV hypertrophy. Although ventricular hypertrophy is typically asymmetric and most commonly involves the interventricular septum, nearly any myocardial segment can be involved ( Fig. 18-8).47–49 Asymmetric septal hypertrophy is more common than diffuse concentric patterns of LV hypertrophy, and thickening of the basal and mid-anterior septal walls are common.48,50,51 Typically, patients will present with a maximal wall thickness measuring between 20 and 30 mm, but in some it may reach 35 to 40 mm. 42 Confusion may arise when the LV hypertrophy is concentric, which may arise from other causes like hypertension or aortic stenosis that result in increased afterload. 40 Hypertrophy of the ventricular septum is most easily appreciated from the midesophageal (ME) four-chamber and long-axis (LAX) echocardiographic views ( Fig. 18-9). Concentric ventricular hypertrophy is best seen in transgastric (TG) short-axis (SAX) images. Oftentimes there will be near-obliteration of the LV cavity during systole when imaged from the TG window. The 2D echocardiographic criteria used to diagnose HCM include maximal wall thickness greater than 15 mm in any myocardial segment that cannot be explained by other causes (confusion may arise when maximum wall thickness is 13-15 mm 40), septal/posterior wall thickness ratio greater than 1.3 in non-hypertensive patients, and septal/posterior wall thickness ratio greater than 1.5 in hypertensive patients. 47
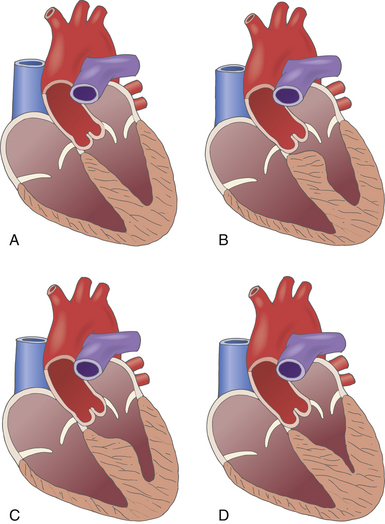
Figure 18-8 Types of hypertrophic obstructive cardiomyopathy. A, A normal heart. B, Hypertrophic disease isolated to basal portion of interventricular septum. C, Hypertrophic disease of midventricular septum resulting in midventricular outflow obstruction. D, Apical hypertrophic disease resulting in small left ventricular cavity and small apical pouch. (Used with permission of Mayo Foundation for Medical Education and Research. All rights reserved.)
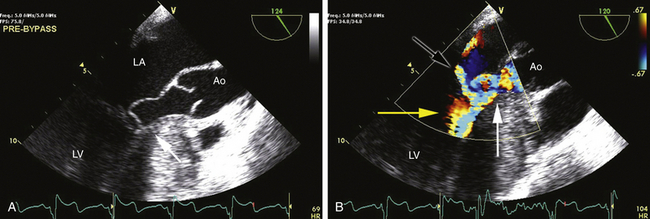
Figure 18-9 A, Typical two-dimensional appearance of hypertrophic obstructive cardiomyopathy. Image is a midesophageal view with a probe angle of 124 degrees. Note hypertrophy of the basal septum (arrow). In this image from midsystole, entire mitral apparatus has been drawn into left ventricular outflow tract (LVOT), causing obstruction to left ventricular emptying and a coaptation defect between mitral leaflets. B, Similar view with color Doppler imaging added. Note typical posteriorly directed jet of mitral regurgitation (black arrow). Color aliasing indicating obstruction to ventricular emptying begins at base of heart just below site of hypertrophy (yellow arrow) and worsens in LVOT, indicating high-velocity turbulent flow (white arrow). These color signals often combine to form the appearance of a Y. Ao, Aorta; LA, left atrium; LV, left ventricle.
Systolic function of the LV in HCM is usually normal or hyperdynamic, with only a small subset of patients having impaired systolic function. If there is severely depressed systolic function, it is often accompanied by LV dilatation and thinning of the ventricular walls. 47
HCM is inherited in an autosomal dominant manner. 41 It may be caused by over 1400 mutations in any of 8 genes; 40 however, three genes that encode proteins of the cardiac sarcomere predominate in frequency. 49 The similarity of the genes accounts for the many different expressions of HCM that resemble one disease entity. The causal mutations of the genes encode the changes in sarcomeric function, leading to hypertrophy and fibrosis beginning as early as puberty. 8 There may be any number of abnormalities generated, such as alterations of protein structure that change interactions or sensitivity to regulators (e.g., calcium), resulting in changes in force or velocity of myocyte contraction. 42 At present, the pathway from gene mutation to clinical expression is still only partially understood. Not all patients who possess a gene for HCM will manifest clinical features of the disease, reflecting incomplete genetic expression. 39 The phenotype appears not only to depend on the mutation but also on other modifier genes and environmental factors.49,52 Patients without a family history of HCM may have sporadic gene mutations or simply a very mild form of the disease. A preclinical diagnosis in an asymptomatic person is possible with gene testing but is not routine, nor can it be used to establish a treatment strategy.
HCM is unique for its range of clinical presentations from infancy to 90 years of age. While major referral centers described a disease with severe symptomatology, many elderly individuals with mild to asymptomatic disease were unaccounted for in past studies, resulting in an annual mortality rate of 3% to 6% per year; more recent studies show it to be closer to 1% per year. 39 Most HCM patients are asymptomatic or have mild symptoms that progress slowly or not at all. 14 If symptoms appear, it is usually in the second or third decade. However, LV hypertrophy can occur at any age and increase or decrease dynamically throughout the person’s life. 39
Symptoms of HCM are nonspecific and include chest pain, palpitations, dyspnea, and syncope. Dyspnea occurs in 90% of patients secondary to diastolic abnormalities that increase filling pressures, causing pulmonary congestion. Syncope occurs in only 20% of patients, but 50% may have presyncopal symptoms. The ECG is abnormal in most individuals (showing increased QRS voltage, ST-segment and T-wave abnormalities, QRS axis shift, and LV hypertrophy with strain pattern) but may be normal in 5% of patients. 42 There is little correlation between ECG voltages and degree of LV hypertrophy. 39 Normal sinus rhythm predominates, but patients with ambulatory monitoring show supraventricular tachycardia (46%) and nonsustained VT (26%). Atrial fibrillation may occur in 25% to 30% of older patients, with serious consequences. 42 A chest roentgenogram may show LA enlargement or be normal.
The most important clinical decision is to identify whether the patient has a nonobstructive or obstructive form of HCM, because management strategy will be largely centered on the presence or absence of symptoms that ensue. Two thirds of patients will have left ventricular outflow tract (LVOT) obstruction. 40 In a recent prospective study of 320 patients with HCM and their gradient, 37% had LVOT obstruction at rest and 33% required provocation. 53 This may add to the unreliability of clinical diagnosis of HCM, because classic physical findings associated with LVOT obstruction are not always present.
Septal hypertrophy and anterior displacement of the papillary muscles and mitral leaflets narrows the LVOT. High-velocity blood flow during ventricular systole creates a drag that mobilizes the mitral valve (particularly the anterior leaflet) into the LVOT, further decreasing the functional LVOT orifice, with the anterior mitral leaflet making contact with the ventricular septum in midsystole, causing abrupt cessation of LV emptying and closure of the aortic valve. Elongation of the mitral leaflets results in coaptation of the body of the leaflets instead of the tips. The part of the anterior leaflet distal to the coaptation is subjected to strong Venturi forces that provoke systolic anterior motion (SAM), mitral septal contact, and ultimately LVOT obstruction ( Fig. 18-10). 49 SAM of the mitral leaflets occurs in roughly 30% to 60% of patients with HCM. The end result of this dynamic process is early closure of the aortic valve due to obstruction to outflow, decreased stroke volume, and MR if the SAM results in a coaptation defect between the mitral leaflets. SAM can occur prior to the opening of the aortic valve by the generation of maximum Venturi effect due to the position of the mitral leaflets on the LVOT, reflecting its importance in SAM compared to traditional explanations. Longitudinal flow in the ventricle may push the mitral valve into the LVOT. 54 SAM of the anterior leaflet may also cause MR unlike that seen with intrinsic structural abnormalities of the valve. Both SAM and LVOT obstruction can be readily appreciated with 2D echocardiographic imaging, an important method of differentiating obstructive from nonobstructive HCM (see Fig. 18-9).
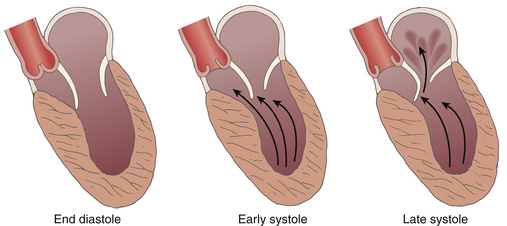
Figure 18-10 Depiction of modern concepts of mechanism of left ventricular outflow tract obstruction in hypertrophic cardiomyopathy. In early systole, abnormal flow around hypertrophied septum pushes mitral valve into outflow tract and results in obstruction and mitral valve regurgitation. (From Ommen SR, Shah PM, Tajik AJ. Left ventricular outflow tract obstruction in hypertrophic cardiomyopathy: past, present and future. Heart. 2008;94:1276-1281.)
The onset and duration of mitral leaflet–septal contact determine the magnitude of the gradient and the degree of MR. The pressure gradient between the aorta and LV is worsened by decreased end-diastolic volume, increased contractility, or decreased aortic outflow resistance ( Fig. 18-11). 55 The cavity of the LV is often very small in those with severe gradients. Doppler echocardiography is essential to defining the presence, location, and severity of LVOT obstruction when present. CWD is helpful for measuring the absolute magnitude of gradient across the LVOT, whereas PWD can define the anatomic location of the obstruction, whether it be in the midventricle or at the base of the heart.
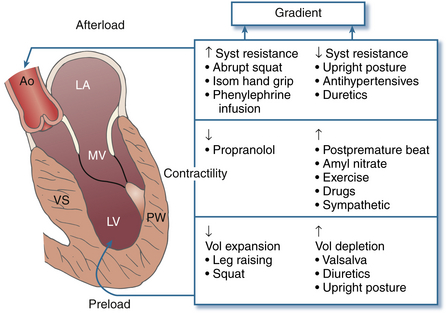
Figure 18-11 Interventions that change outflow gradient in hypertrophic cardiomyopathy (HCM), with resultant change in intensity of systolic murmur in HCM. Outflow gradient is affected by changes in afterload, preload, and contractility. Ao, Aorta; LA, left atrium; LV, left ventricle; MV, mitral valve; PW, posterior wall; VS, ventricular septum. (From Giuliani ER, Fuster V, Gersh BJ, et al. Cardiology: Fundamentals and Practice. St. Louis: Mosby; 1991.)
TEE CWD may be used from deep TG, ME four-chamber, or ME LAX views to measure the maximum instantaneous gradient ( Fig. 18-12). The typical CWD appearance of the LVOT signal is that of a high-velocity “dagger-shaped” configuration. Particularly when imaging from the esophagus, the LVOT signal is relatively easy to confuse with that of MR. Velocities from both signals may be similar, but the signal due to MR tends to be more symmetric and rounded, lacking the dagger-shaped appearance of the LVOT signal ( Fig. 18-13).
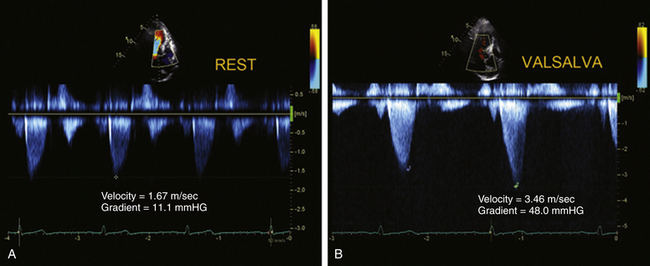
Figure 18-12 Continuous wave Doppler spectra obtained from apex, demonstrating dynamic left ventricular outflow tract obstruction. Note typical late-peaking configuration resembling a dagger or ski slope (A and B). The baseline (A) velocity is 1.67 m/s, corresponding to the peak LV outflow tract of 11.1 mmHg (4 × 1.672). With the Valsalva maneuver (B), the velocity increased to 3.46 m/s, corresponding to a gradient of 48 mmHg.
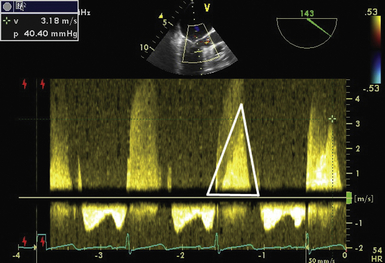
Figure 18-13 The probe is in the midesophagus with an angle of 143 degrees. A continuous wave Doppler signal has been placed through mitral valve and left ventricular outflow tract. Note in the Doppler signal tracing that denser signal from outflow tract is seen within lighter-shaded, higher-velocity signal of mitral regurgitation (outflow tract signal outlined by white triangle). Signal has a steep upslope due to rapidly increasing velocity as obstruction to outflow worsens throughout systole, giving signal the appearance of a “dagger.” In the subsequent cardiac cycle, echocardiographer is able to measure the gradient through the outflow tract even though it is confined within mitral regurgitation signal. Velocity of outflow tract signal is 3.18 m/s, yielding a gradient of 40.4 mmHg.
Color Doppler imaging reveals a multitude of subjective findings in patients with obstructive HCM. Typical findings in patients with obstructive disease include flow acceleration in midventricle or at the base of the LV as blood flow begins to accelerate upon approaching the LVOT. Aliasing is seen in the LVOT as blood flow reaches maximum acceleration due to LVOT obstruction, and the jet of MR is also seen (see Fig. 18-9). Indeed, the classic description of color Doppler imaging in obstructive HCM is that of acceleration → obstruction (to LVOT flow) → leak (MR). Along with PWD findings, the point of flow acceleration seen with color Doppler imaging can give valuable insight as to the point of obstruction to LV emptying. This finding has important implications when planning the surgical approach to septal myectomy.
Dynamic pressure gradients do not necessarily correlate with symptoms of HCM. Significant functional limitation, disability, and sudden death may all occur without a gradient, but gradients exceeding 30 mmHg are usually of physiologic and prognostic importance in HCM patients. 49 Two thirds of individuals with LVOT obstruction will become severely symptomatic, and there is 10% mortality within 4 years of diagnosis. LVOT obstruction is an independent predictor of death in HCM. 56
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


