Cardiac Tumors
Bo Yang
Himanshu J. Patel
Francis D. Pagani
Richard L. Prager
HISTORICAL BACKGROUND
Realdo Columbus first described a cardiac tumor as an anatomic finding in Padua, Italy, in 1559. Centuries later, in 1931, the first classification system similar to what is in current use was reported by Yater. In his monograph, he reported nine cases of primary cardiac tumors from pathologic examination. However, it was not until 1934 that the first antemortem diagnosis of a cardiac tumor (sarcoma) was made by Barnes, who used electrocardiography and a biopsy of a lymph node. Twelve years later, Mahaim’s classic paper described over 400 cardiac tumors.
The era of operative treatment of cardiac neoplasms was ushered in by Beck in 1936, when he successfully removed a teratoma located on the right ventricular (RV) surface. Bahnson is credited with the removal of the first right atrial myxoma with inflow occlusion, but the patient died postoperatively on day 24. Following the advent of cardiopulmonary bypass in 1953, left-sided intracardiac neoplasms were successfully removed first by Crafoord in Sweden in 1954. By 1964, the removal of 60 intracardiac neoplasms had been reported. The addition of cardiac echocardiography allowed easier antemortem diagnosis, and thus resulted in an increase in the number of tumor resections.
EPIDEMIOLOGY AND CLASSIFICATION
Cardiac neoplasms can be divided into primary and secondary types. Primary tumors are typically more common, but the overall incidence is still quite low, at 0.15% to 0.2% in autopsy series. Most primary cardiac tumors are benign (70%; Table 68.1). More than half of the primary cardiac neoplasms in adults are myxomas.
Malignant primary cardiac tumors (Table 68.2) are more frequently seen in adults than in children. Of malignant cardiac tumors, metastatic malignancies (Table 68.3) comprise the majority of those noted. Virtually every neoplasm has been shown to metastasize to the heart. The most frequent primary malignancies are leukemic neoplasms (54%), melanoma (34%), and bronchogenic carcinoma (10%), with others including sarcoma, breast, and esophageal carcinoma. Primary cardiac malignancies are uncommon, and most frequently are sarcomas.
MYXOMAS
Myxomas are the most frequently seen adult cardiac neoplasm. They usually occur sporadically but have been reported in autosomal-dominant inherited forms in 5% of cases. The typical sporadic tumor is seen in women aged 30 to 60 years and is solitary in nature. The familial form is more likely seen in younger patients, often male, and multicentric in nature. The two types can be differentiated on the basis of DNA ploidy, with the familial type having an abnormal ploidy. Carney complex is an autosomal X-linked inheritance characterized by cardiac myxoma, cutaneous pigmentous lentigines, and primary pigmented adrenocortical disease with hypercortisolism.
Myxomas are typically located in the atria (most commonly on the limbus of the fossa ovalis) (Fig. 68.1A) but can arise in the ventricles. Multiple myxomas with involvement of different heart chambers have been reported but are very rare. The tumors are variable in their gross pathologic appearance (Fig. 68.1B), and can be papillary or smooth, pedunculated, or sessile, but are often quite friable. They are usually white or yellow and may be covered with thrombus. On cut sections, they frequently contain areas of hemorrhage. They are typically 5 to 6 cm in size but have been reported to reach up to 15 cm. Histopathologically, they have an acid mucopolysaccharide matrix and contain smooth muscle cells, capillaries, and reticulocytes. Calcification is reported more frequently in right atrial myxomas. These tumors typically grow outward into the cardiac chambers and rarely invade into the walls of the heart. However, those tumors that are biatrial in location are thought to be a result of bidirectional growth of the myxoma because they are usually attached to the same point on the atrial wall. Usually, they are limited to the subendocardial region at their base. The subendocardial multipotential mesenchymal cell is considered the precursor cell for myxoma, thus accounting for the variable cell types seen in the tumor.
The natural history of myxomas is of rapid growth. Although these tumors are considered to be benign, there have been reports of extensive local extension, as well as metastatic spread. The familial type is more likely to be recurring and more aggressive.
The clinical presentation patterns of myxomas relate to their potential to cause obstruction (congestive heart failure, atrial fibrillation, fatigue, and syncope), embolization, and constitutional symptoms (myalgias, fevers, arthralgias, and weakness). Occasionally, they can present with evidence of infection, with a syndrome not unlike infective endocarditis. Physical findings can include signs of right- or left-sided congestive failure, the early diagnostic “tumor plop,” or diastolic rumbles.
The workup of a suspected myxoma includes echocardiography as the most useful test for diagnosis. Although surface echocardiograms can identify the pathology in most cases, the transesophageal echocardiogram (Fig. 68.1C) gives the best images and provides details regarding the location and attachment areas for tumors even as small as 2 mm. This aids in planning the operative approach. We also request left heart catheterizations to aid in the diagnosis of coronary disease in patients older than the age of 45 years or even younger if they have significant risk factors or symptoms. Computed tomography (CT) and magnetic resonance imaging (MRI) are rarely utilized at our center for suspected myxomas because in the setting of a myxoma, they typically do not add additional information. However, if there is suspicion that the
tumor may not be a myxoma, cardiac-gated CT scans or cardiac-gated MRI may better delineate the extent of involvement of adjacent structures by the tumor. It is important to exclude endocarditis as a cause of cardiac mass.
tumor may not be a myxoma, cardiac-gated CT scans or cardiac-gated MRI may better delineate the extent of involvement of adjacent structures by the tumor. It is important to exclude endocarditis as a cause of cardiac mass.
Table 68.1 Benign Cardiac Neoplasms | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
Table 68.2 Malignant Primary Cardiac Neoplasms | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
Table 68.3 Metastatic Cardiac Neoplasms | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
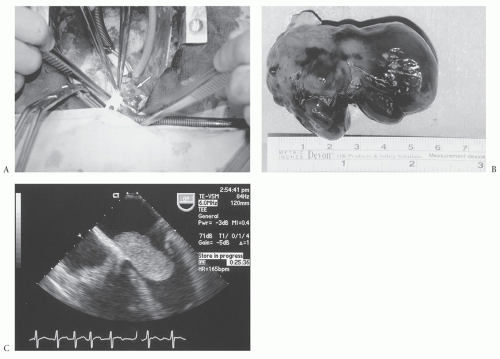 Fig. 68.1. (A) Typical operative approach using median sternotomy and cardiopulmonary bypass using a bicaval inflow technique. The exposure of the tumor is via a biatrial approach, and the tumor (white arrow) is readily delivered via the left atriotomy. (B) Gross pathologic appearance of this myxoma, which consists of large, mottled-tan hemorrhagic tissue, somewhat gelatinous and myxoid, measuring 6 cm in maximal dimension. (C) Transesophageal echocardiogram of this tumor in vivo. Note the “ball-valve” obstruction of the mitral valve caused by the tumor. The attachment is to the left atrial wall, along the fossa ovalis. |
The indication for operation in patients with a myxoma is the presence of one. Given the risk of embolization (8% to 10%) or obstruction, we typically perform the operation in an urgent manner following the establishment of diagnosis. These patients are usually operated on during the same hospitalization. Initial attempts are made to diurese the patients while obtaining necessary workup, but the surgery is typically not delayed. Anticoagulation with heparin is important to
institute during the time of evaluation because it is felt to decrease the risk of embolization.
institute during the time of evaluation because it is felt to decrease the risk of embolization.



