Atherothrombosis: Disease Burden, Activity, and Vulnerability: Introduction
Atherosclerosis with thrombosis superimposed, atherothrombosis, is the main cause of myocardial infarction, coronary death, heart failure, and large-artery stroke.1-3 The development of thrombosis-prone atherosclerotic plaques, also known as high-risk or vulnerable plaques,4 in the coronary and carotid arteries is the leading cause of death and severe disability, not only in affluent countries, but also worldwide.5,6
Causal and modifiable risk factors for atherosclerotic cardiovascular disease (CVD) are well known (smoking, dyslipidemia, high blood pressure, diabetes, etc) and account for most heart attacks in both sexes.7 However, for unknown reasons, the individual susceptibility to these risk factors varies greatly, and consequently, their predictive value is limited.8,9 Most first heart attacks occur among people with average or only slightly elevated risk factor levels,10-13 and recurrent events still occur despite lowering of these levels,14,15 indicating that we need both better detection and better treatment of those who are destined for a heart attack. Better detection of at-risk individuals may be achieved by visualizing the diseased arterial wall rather than just assessing risk factors for getting the disease.16,17 The prospect for atherosclerosis-based risk assessment will be discussed in the concluding section.
Atherogenesis and Plaque Development
Atherosclerosis is a chronic, lipid-driven inflammatory disease of the arterial wall leading to multifocal plaque development,18-20 predominantly at predilection sites characterized by low and oscillatory endothelial shear stress (bifurcations, inner wall of curvatures) and preexisting intimal thickenings.21,22 The speed of disease progression varies greatly, but it usually takes decades to develop the advanced atherosclerotic lesions responsible for clinical disease. Plaques are very heterogeneous in size and composition, even plaques located next to each other and exposed for the same systemic risk factors (Fig. 52–1). Most plaques remain asymptomatic (subclinical disease), some become obstructive (stable angina), and a few, if any, become vulnerable and lead to atherothrombotic events such as a fatal heart attack or a disabling stroke.1-4
Figure 52–1.
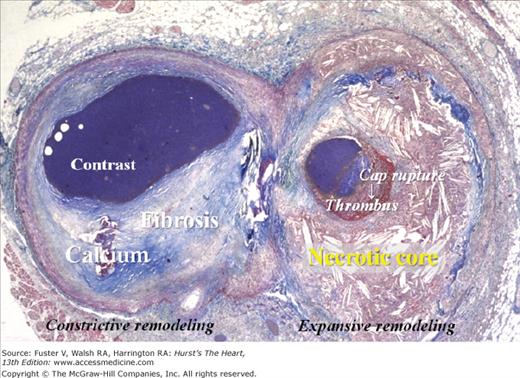
Heterogeneity of atherosclerotic plaques. Cross-section of a coronary artery cut just distal to a bifurcation. The plaque to the left (circumflex branch) is fibrotic with a dense calcification, whereas the plaque to the right (marginal branch) contains a large lipid-rich necrotic core covered by a thin fibrous cap that is disrupted with mural thrombosis. The lumen contains contrast medium injected postmortem. Trichrome, staining collagen blue and thrombus red.
The endothelium, located at the interface between the blood and the arterial wall, plays a key role in the development of atherosclerosis.22 In lesion-prone areas, apoprotein B–containing lipoproteins extravasate into intima, where they are retained and modified (eg, oxidized) into cytotoxic, proinflammatory, chemotaxic, and proatherogenic molecules.23 The overlying endothelium becomes activated and mediates transendothelial trafficking of leukocytes, especially monocytes but also T cells and a few mast cells, primarily via upregulated adhesion molecules.18,19
Monocyte-derived macrophages play a key role in the initiation, progression, and destabilization of atherosclerotic plaques.18-20,24,25 Some macrophages perform beneficial scavenger functions, cleaning up intima for toxic lipid-rich waste products; others are destructive and accelerate the development of a destabilizing necrotic core and a rupture-prone fibrous cap as described below. Consequently, macrophage number and function within atherosclerotic plaques are obvious putative markers of disease activity. However, because atherosclerosis is a protracted and smoldering inflammatory disease in which the advanced and potentially symptomatic lesions are dominated by fibrosis, extracellular lipids, necrosis, and calcification (Fig. 52–2),26 the macrophage density is usually low, and macrophages rarely occupy more than a small percentage of the plaque.27 In fact, atherosclerosis is a smoldering inflammatory disease without much inflammation.
Figure 52–2.
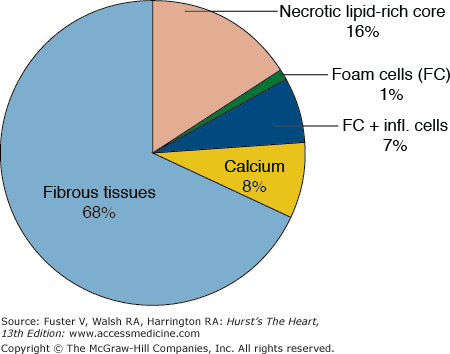
Average composition of advanced coronary plaque. Pie diagram illustrating the average composition of advanced atherosclerotic plaques (>75% stenosis by histology) in the coronary artery tree in fatal myocardial infarction.26 Hypocellular tissues (fibrosis, calcium, and necrosis) constitute by far the most voluminous plaque components.
When intimal damage occurs, vascular smooth muscle cells (SMCs) modulate into a synthetic phenotype and perform fibroblast-like functions. They migrate, proliferate, and synthesize extracellular matrix, including collagen, and thus confer stability to plaques.28 Analogous to scar contraction, collagen-rich plaques tend to contract and aggravate luminal narrowing. If the vascular SMCs vanish, healing and repair are compromised and the macrophage-mediated destructive processes will prevail.
During atherogenesis, some cells proliferate and others die by apoptosis or necrosis. The rate of cell proliferation is low and predominantly confined to inflammatory cells.29 Apoptosis is also most common in inflammatory cells, but its rate is more difficult to determine because of defective clearance of dead cells in advanced lesions.30-32 Death of macrophage foam cells and smooth muscle cells are believed to play important pathogenetic roles in the formation of a necrotic core and thinning of the overlying fibrous cap, respectively.25,30-32
Subsets of mononuclear cells in the blood, termed endothelial progenitor cells and smooth muscle progenitor cells, form colonies in cell culture with endothelial or smooth muscle cell-like features, respectively.33,34 These circulating cells, assumed to originate solely or mainly from the bone marrow, have been claimed to home and differentiate into bona fide endothelial and smooth muscle cells in atherosclerotic lesions and thus promote healing and repair.35,36 However, recent data question this theory. First, the putative endothelial progenitor cells identified in clinical studies have been shown to give rise to colonies composed of inflammatory and immune cells, rather than endothelial cells, in vitro.37,38 Second, new mouse studies using improved techniques to track endothelial and smooth muscle cell origin in atherosclerotic plaques have found that both cell types are derived from local sources with rare, if any, contributions from the blood.39-41 Thus, rather than differentiating into endothelial or smooth muscle cells, the so-called progenitor cells in the blood might influence the progression of atherosclerosis by paracrine signaling, as appears to be the case for neovascularization (angiogenesis).42
Vulnerable and Thrombosed Plaques
The great majority of symptomatic coronary thrombi (~75%) are caused by plaque rupture.20 Plaque rupture with mural thrombosis (with or without plaque hemorrhage) is also a common cause of episodic but asymptomatic progression to severe stenosis.4,43,44 The remaining thrombi are caused by less well-defined mechanisms, of which so-called plaque erosion is the most common type.4 Plaque rupture is a more frequent cause of coronary thrombosis in men (~80%) than in women (~60%) but, except for sex and menopause, no other risk factors have consistently been connected with a particular mechanism of thrombosis.20 By inference, there are two major types of vulnerable plaques, rupture-prone and erosion-prone, that are presumed to look like the corresponding thrombosed plaques, just without rupture and thrombosis.4,45
The prototype of a presumed rupture-prone plaque contains a large and soft lipid-rich necrotic core covered by a thin and inflamed fibrous cap (Fig. 52–3).25,45 Associated features include big plaque size, expansive remodeling mitigating luminal obstruction (mild stenosis by angiography), neovascularization (angiogenesis), plaque hemorrhage, adventitial inflammation, and a spotty pattern of calcifications (Fig. 52–4).20 Although the macrophage density in ruptured caps is high,25,45 whole-plaque macrophage density rarely exceeds a small percent because ruptured caps are tiny (Fig. 52–5).
Figure 52–3.
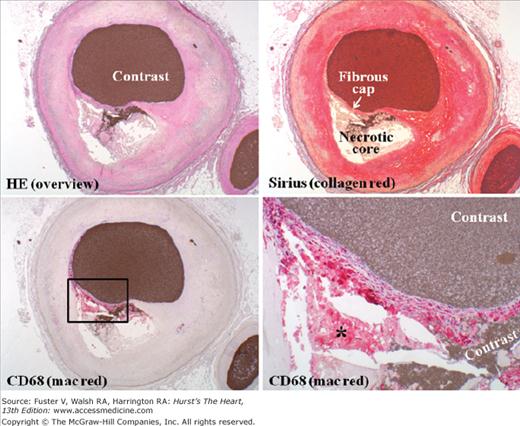
Inflamed thin-cap fibroatheroma (TCFA). Coronary TCFA composed predominantly of hypocellular fibrous tissue and acellular necrotic core. Penetration of contrast medium injected postmortem from the lumen into the necrotic core reveals fibrous cap rupture. Immunohistochemistry visualizes abundant macrophages (mac) within the thin fibrous cap but virtually nowhere else. Beneath the cap, dead macrophages without nuclei are seen within the necrotic core (asterisk). The cap contains very few smooth muscle cells (not shown). HE, hematoxylin and eosin.
Figure 52–4.
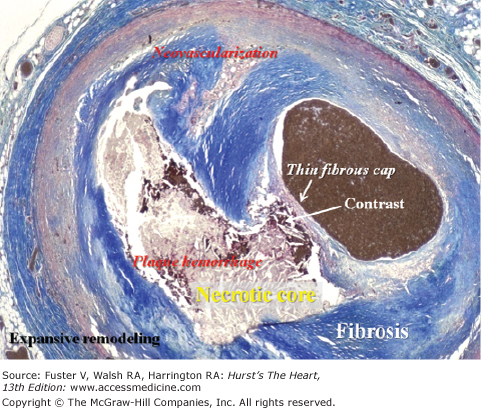
Coronary thin-cap fibroatheroma (TCFA). A TCFA containing a soft and destabilizing necrotic core devoid of supporting collagen and separated from the lumen only by a thin fibrous cap. Such a plaque is presumed to be rupture-prone and, if correct, constitutes the most common type of vulnerable plaques. In this case, postmortem contrast injection provided proof of vulnerability; the contrast has penetrated from the lumen into the necrotic core, indicating the presence of a disrupted fibrous cap. The contrast followed a path bordered by extravasated erythrocytes (plaque hemorrhage). Trichrome, staining collagen blue (necrotic core not stained).
Figure 52–5.
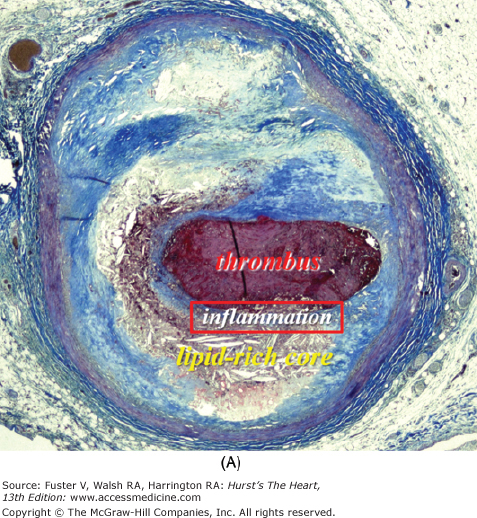
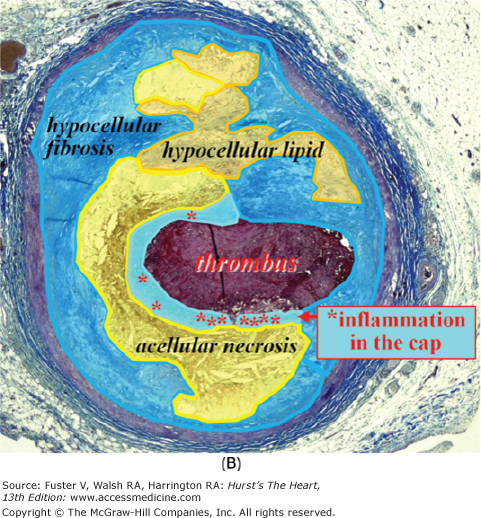
Ruptured coronary plaque with thrombosis. The thin cap is heavily inflamed, whereas the remainder of the plaque is not. Macrophages (asterisk) cluster in the cap next to the rupture site. The majority of the plaque consists of acellular lipid-rich core and hypocellular fibrosis and lipid pools.
Vulnerable plaques of the erosion-prone type are heterogeneous and defined only by their fate (thrombosis, mostly mural) (Fig. 52–6).4 The surface endothelium is missing, but, whether it vanished before or after thrombosis remains unknown. No distinct morphologic features have been identified, but in general, eroded plaques with thrombosis are scarcely calcified, rarely associated with expansive remodeling, and only sparsely inflamed.4 So, irrespective of plaque type, it is a misconception that vulnerable plaques are heavily inflamed.
Figure 52–6.
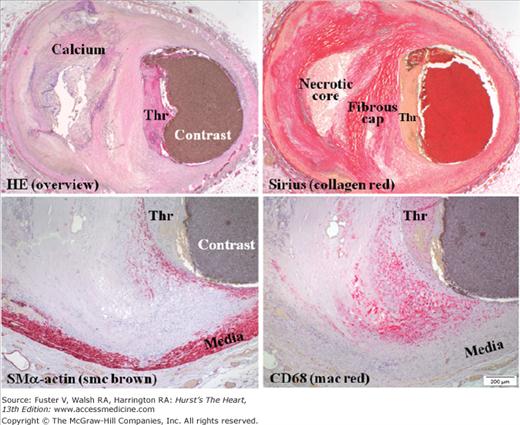
Thrombosis not caused by plaque rupture. Coronary artery containing a nonruptured plaque with mural thrombosis (Thr), so-called plaque erosion. The endothelium between the plaque and the thrombus is missing, but no other morphologic features characterize this type of thrombosed plaques and their vulnerable precursors. The plaque is rich in smooth muscle cells (smc), but not macrophages (mac), just beneath the thrombus. Focal mac accumulation is seen at the shoulder region of the plaque but not located within a thin cap (ie, this plaque morphology is not permissive for plaque rupture). HE, hematoxylin and eosin; SMα, smooth muscle α.
Vulnerable plaques, plaque rupture, and thrombosed plaques tend to cluster in hot spots within the proximal segments of the major coronary arteries,46-48 and rarely more than one or a few such lesions exist simultaneously.49,50 The natural history of vulnerable plaques such as speed of development, lifetime (persistency), and fate, is, however, unknown.
Determinants of Plaque Vulnerability
Plaque rupture requires the presence of a lipid-rich necrotic core covered by a thin fibrous cap. The size of the necrotic core and the thickness of the fibrous cap appear to be the two major structural determinants of vulnerability (Fig. 52–7).
Figure 52–7.
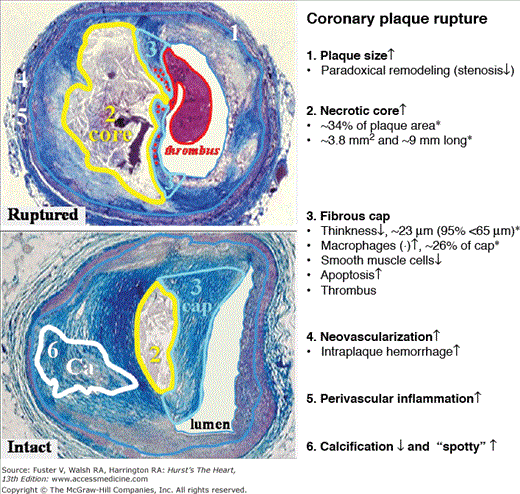
Plaque rupture and determinants of vulnerability. For comparison, a ruptured plaque with thrombus (top) and an intact and stable plaque (bottom) are depicted, and vulnerable plaque features are listed to the right. By inference, vulnerable plaques of the rupture-prone type are presumed to look like plaque rupture except for an intact cap without thrombus. Data were derived from Kolodgie et al.56
During atherogenesis, atherogenic lipoproteins are retained within intima, modified, and accumulate, predominantly deeply in the abluminal part of intima.51,52 Some of these pools of lipids seem to attract macrophages that secrete proteolytic enzymes and engulf lipid until they die, leaving behind a soft and destabilizing lipid-rich cavity containing cholesterol crystals and devoid of supporting collagen and cells, the necrotic core.25 Such a plaque is called an atheroma or fibroatheroma.53,54 Later on, extravasation of erythrocytes into the necrotic core may expand it (see Neovascularization and Plaque Hemorrhage later).
The fibrocellular part of the plaque located between the necrotic core and the lumen is called the fibrous cap. It is extremely thin in coronary plaque rupture.25 Assessed by microscopic examination postmortem, ruptured caps were usually <65 μm thick.54 Assessed by optical coherence tomography in vivo, the mean thickness was only 49 μm.55 If the fibrous cap is thin, the plaque is called a thin-cap fibroatheroma (TCFA).54,56 In TCFA, the necrotic core occupies approximately 23% of plaque area.56 Thin fibrous caps are usually heavily inflamed (macrophage density ~14%), particularly those that have ruptured (macrophage density ~26%),56 but because they are thin, their ability to accommodate macrophages are limited (see Fig. 52–5). Apoptosis is common at the site of fibrous cap rupture, usually confined to macrophages because the vascular SMCs already have vanished when rupture occurs.31,56 With their ability to synthesize extracellular matrix, including collagen, SMC apoptosis is associated with impaired healing and repair, increasing the risk of plaque rupture.
Atherosclerosis is an innate inflammatory disease in which smoldering inflammatory activity is not confined to just a few atherosclerotic lesions but is present, more or less, in all such lesions throughout the body.18,19 In contrast, vulnerable plaques are relatively rare,49 and inflammation may play a causal role in plaque rupture only if located within a thin fibrous cap (ie, the microstructure of the plaque needs to be permissive for rupture). Thus, although plaque inflammation may be useful as a marker of disease activity, it is probably not useful as a standalone marker for plaque vulnerability (see Figs. 52–5 and 52–7).
During atherogenesis, the artery tends to remodel in such a way that the luminal obstruction caused by some plaques is attenuated (expansive remodeling) and by others accentuated (constrictive remodeling). Although vulnerable plaques of the rupture-prone type (TCFA) usually are big, they often appear nonobstructive by angiography because of local expansive remodeling and extension of the disease to the adjacent reference segments judged to be normal by angiography.25,57,58 In contrast, plaques responsible for stable angina usually are smaller but nevertheless are often associated with more severe luminal narrowing by angiography because of concomitant constrictive remodeling.59 The reasons for the different modes of remodeling remain to be defined, but recent clinical observations indicate that diabetes is accompanied by inadequate compensatory remodeling.60
Angiogenesis and inflammation often coexist at the base of advanced plaques.61 The new microvessels rarely originate from the lumen but usually originate from vasa vasorum in adventitia.62
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


