Atherosclerosis: Epidemiology and Pathophysiology
Seung Uk Lee
Joanna J. Wykrzykowska
Roger J. Laham
Atherosclerosis, a progressive disease characterized by the accumulation of lipids and fibrous elements in the large arteries, is the leading cause of death and disability in the United States and other industrialized nations (1). Cardiovascular atherosclerotic disease with its resultant ischemic heart disease, stroke, and peripheral vascular disease is responsible for approximately 50% of all deaths in the United States (2,3). The clinical manifestations of atherosclerotic disease are myriad often coexisting conditions. These include chronic conditions such as stable angina pectoris, peripheral vascular disease, and aneurysmal dilation as well as acute life-threatening conditions such as stroke, myocardial infarction, limb-threatening ischemia, and aneurysm rupture. However, the prevalence of atherosclerotic disease far exceeds the incidence of its adverse events. This raises a concern that with the aging population, atherosclerotic disease will become more manifest.
Significant improvements in the treatment of atherosclerotic heart, cerebral, and peripheral vascular disease have been made. These have included risk factor modifications, antiplatelet and anticoagulant, lipid-lowering agents (“statins”), and antihypertensive medications as well as procedures and devices (such as percutaneous and surgical revascularization). Despite this, the prevalence of atherosclerotic disease continues to rise, and its resultant events continue to burden the health care system. This is partly due to the progressive nature of this disorder, improved survival with treatment, increasing age of the population, and an incomplete understanding of the underlying biology.
Recent advances in our understanding of the fundamental cellular and molecular pathways involved have established an essential role for inflammation, vessel wall injury, and angiogenesis in the various stages of atherosclerosis.
The inflammatory processes result from the interaction between modified lipoprotein, macrophages, and T-cells, among others. This mechanism provides a more direct link between the known risk factors and the pathogenesis of the disease.
Thus, atherosclerosis, formerly considered a degenerative lipid storage disease, is currently viewed as a form of chronic inflammation that can result in acute plaque rupture, thrombosis, and embolization (4,5). This chapter will focus mainly on the epidemiology, biology, and clinical manifestations of atherosclerosis, with a special emphasis on the emerging dominant role for inflammation in this disease.
Atherosclerosis Statistics
In 2002, the estimated prevalence of total cardiovascular disease (CVD), coronary heart disease (CHD), and congestive heart failure (CHF) in the U.S. population was 70 million, 13 million, and 4.9 million, respectively. The estimated prevalence of myocardial infarction and stroke, the
most serious acute complication of atherosclerotic disease, was 7.1 and 5.4 million, respectively. CVD accounted for 38% of all deaths, or 1 of every 2.6 deaths, in 2002. CHD caused 494,382 deaths, or 1 of every 5 deaths, in the United States. Despite marked improvement in therapeutic options and a significant decline in the death rate of CVD from 1992 to 2002, the actual number of deaths increased 0.8%. According to current statistics and estimates, 865,000 new and recurrent myocardial infarctions and 700,000 new and recurrent stroke attacks are expected annually.
most serious acute complication of atherosclerotic disease, was 7.1 and 5.4 million, respectively. CVD accounted for 38% of all deaths, or 1 of every 2.6 deaths, in 2002. CHD caused 494,382 deaths, or 1 of every 5 deaths, in the United States. Despite marked improvement in therapeutic options and a significant decline in the death rate of CVD from 1992 to 2002, the actual number of deaths increased 0.8%. According to current statistics and estimates, 865,000 new and recurrent myocardial infarctions and 700,000 new and recurrent stroke attacks are expected annually.
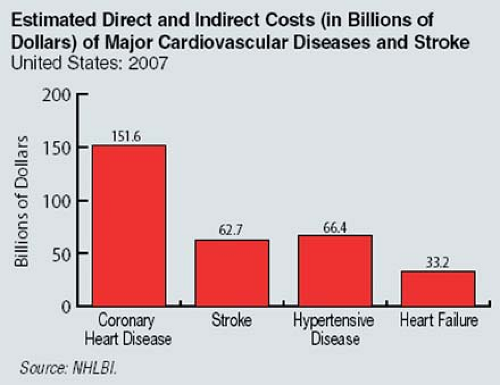 Figure 7-1 Estimated direct and indirect costs (in billions of dollars) of cardiovascular diseases and stroke in the United States in 2007. (Reprinted from Heart Disease and Stroke Statistics—2007 Update, American Heart Association.) |
Furthermore, the economic impact of atherosclerotic disease is staggering. In the United States, costs related to stroke are expected to reach an estimated $62.7 billion in 2007; CHD is projected to cost an estimated $151.6 billion in direct and indirect costs in 2007 (Fig. 7-1).
Risk Factors for Atherosclerosis
Epidemiological studies (both experimental and observational) over the last 50 years have identified several risk factors for the development of atherosclerosis. These can be grouped into factors with an important genetic component and factors with a largely environmental component (Table 7-1). It is important to emphasize that the factors reported are the ones that were investigated, and it is likely that new risk factors will be identified with a better understanding of the biology of atherosclerosis. Large, prospective, community-based observational studies confirmed suspected links between suggested factors and cardiovascular risk. A variety of factors, often acting in concert, have been shown to be associated with an increased risk of atherosclerotic plaques in coronary arteries and other arterial beds (1,6,7).
Table 7.1 Risk Factors Associated with Atherosclerosis | |
|---|---|
|
As portrayed in the Framingham Heart Study, which has been ongoing for decades, the different coronary risk factors have an additive effect on the likelihood of developing coronary heart disease (8). The major risk factors and predisposing conditions include dyslipidemia, tobacco use, diabetes mellitus, hypertension, and genetic factors (family history). Metabolic conditions, infectious agents, and psychiatric conditions can each contribute to the risk of developing atherosclerosis.
Dyslipidemia
Elevated levels of serum cholesterol are important in the development of atherosclerosis in humans and experimental animals even in the absence of other known risk factors. High-fat, high-cholesterol diets are usually required for the development of atherosclerosis in experimental animals (9). Epidemiologic studies have demonstrated a continuous, graded relationship between total serum cholesterol and coronary atherosclerotic risk (10). There are five major groups of lipoproteins that may
be separated by ultracentrifugation: the chylomicrons, very-low-density lipoproteins (VLDL), low-density lipoproteins (LDL), intermediate-density lipoprotens (IDL), and high-density lipoproteins (HDL).
be separated by ultracentrifugation: the chylomicrons, very-low-density lipoproteins (VLDL), low-density lipoproteins (LDL), intermediate-density lipoprotens (IDL), and high-density lipoproteins (HDL).
The major plasma lipids are not water soluble, and they do not circulate in a free form in the blood. Instead, they are complexed and bound to a specific group of protein carriers called apolipoproteins, which are found on the surface of the lipoproteins.
Much of the progress in understanding atherosclerosis over the past several decades has depended on the lipid hypothesis, which links cholesterol with heart disease and atherosclerosis. LDL cholesterol undoubtedly contributes to atherosclerosis in many cases and may indeed constitute a permissive factor for atherogenesis.
Reduction of LDL cholesterol by differing modalities, such as diet, several kinds of lipid-lowering agents, ilial bypass, and LDL apheresis, are associated with a reduction in atherosclerosis progression (11,12,13,14,15). It is clear that serum cholesterol, in particular LDL, may initiate the pathological processes of atherosclerosis.
Lipid-lowering strategies, particularly using the statin class of drugs (3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors, an enzyme essential in cholesterol synthetic pathway), promote features of experimental atheroma associated with plaque stability. These features include a reduction in macrophage number, a decrease in expression of matrix metalloproteinases (MMP), proinflammatory cytokines, leukocyte adhesion molecules, and decrease in production of reactive oxygen molecules. All of these steps are essential for progression of atherosclerosis. Statins also increase interstitial collagen content and smooth muscle maturation (16,17). They have been shown to reduce cardiovascular mortality and may stabilize the plaque (18,19,20,21,22,23,24,25,26,27).
Several large, randomized, controlled trials have established the effect of cholesterol-lowering therapy on the risk of death and cardiovascular events across a wide range of cholesterol levels (with or without history of CHD) (21,22,23,24,25,26). Patients with an acute coronary syndrome benefit from an intensive lipid-lowering (statin) regimen that in turn helps stop the progression of atherosclerosis (27,28). Several statin trials have shown that reduction in LDL cholesterol is associated with a reduced incidence of stroke (29,30,31,32). Therefore, dyslipidemias are an important contributor to the pathophysiology of atherosclerosis.
However, most patients with proven coronary artery disease have “average” levels of cholesterol (33), and statins slow but do not stop the progression of atherosclerosis, suggesting that additional risk factors may be as important.
Tobacco Use
Smoking is strongly associated with atherosclerosis (34,35,36,37,38,39). Between 1995 and 1999, an average of 442,398 Americans died yearly of smoking-related illnesses, with 33.5% of these deaths being cardiovascular. Approximately 90% of peripheral vascular disease in the nondiabetic population can be attributed to cigarette smoking, as can 50% of aortic aneurysms.
Smoking is also a powerful risk factor for CHD (40,41). The global INTERHEART study showed that smoking was a major contributor to 36% of first myocardial infarction (42). The risk of recurrent infarction in a study of smokers fell by 50% within 1 year of smoking cessation and normalized to that of nonsmokers within 2 years (9,43). Moreover, current smoking is a powerful independent predictor of sudden cardiac death in patients with CHD (43).
Cigarette smoking increases inflammatory markers, endothelial dysfunction (ED), platelet aggregation, leukocyte recruitments, and thrombosis in the atheromatous plaque (34,35,38,39). The risk of smoking is amplified by other risk factors, such as diabetes, hypertension, and elevated serum lipid levels (44). The benefits of smoking cessation are evident regardless of how long or how much the patient has previously smoked and should be emphasized in every patient.
Diabetes Mellitus
Diabetes mellitus affects 150 million people worldwide, and the number of diabetics in the United States approaches 20 million. The prevalence of diabetes among adults in the United States has increased by 40% in the past decade and is expected to increase by 165% between 2000 and 2050 (45,46,47). Mortality from myocardial infarction in patients with diabetes is markedly higher than in nondiabetic patients (48,49). Patients with diabetes but without previous myocardial infarction carry the same level of risk for subsequent acute coronary events as nondiabetic patients with previous myocardial infarction. Diabetes worsens early and late outcomes in acute coronary syndromes and long-term prognosis after myocardial infarction (50,51,52,53,54,55,56). Thus, the Adult Treatment Panel III of the National Cholesterol Education Program mandates aggressive antiatherosclerotic therapy in all diabetics (50).
Patients with diabetes have a two- to fourfold increase in the rate of peripheral artery disease (57). Diabetes is associated with arterial bruits, loss of pulse, arterial occlusion, amputation risk, and intermittent claudication (57,58,59,60,61,62,63).
Diabetes also adversely affects the cerebral circulation, with more extracranial atherosclerosis and a fivefold increase in the prevalence of calcified carotid atheroma (64,65). The risk of stroke is increased 150% to 400% with diabetes and is directly related to worsening glycemic control (66,67,68,69). In the Multiple Risk Factor Intervention Trial (MRFIT), subjects taking medications for diabetes were three times more likely to develop a stroke (70).
Diabetes also affects the risk of stroke among younger patients (71,72) and affects stroke outcomes. There is a threefold increase in the risk of stroke-related dementia, the risk of recurrence is doubled, and there is an increase in total and stroke-related mortality (73,74,75). The abnormal metabolic state in diabetes causes vascular dysfunction and atherosclerosis through several mechanisms, including chronic hyperglycemia, dyslipidemia, and insulin resistance. Diabetes mellitus alters the function of multiple cell types, including endothelial cells, smooth muscle cells, and platelets (76). Hyperglycemia impairs endothelial function, augments vasoconstriction, increases inflammation, and promotes thrombosis. By decreasing nitric oxide (NO) and increasing endothelin-1 and angiotensin II concentrations, hyperglycemia increases vascular tone and vascular smooth muscle cell proliferation and migration (77,78,79).
Increase in vascular tone in hyperglycemia is mediated by activation of protein kinase-C and nuclear factor [kappa] B in vascular smooth muscle cells and endothelial cells. This leads in turn to increased production of O2- and contributes to the oxidant-rich milieu (80). Arterial vascular smooth muscle cells cultured from patients with type 2 diabetes demonstrate enhanced migration (81). LDL that has undergone nonenzymatic glycation induces vascular smooth muscle cell migration in vitro, while oxidized glycated LDL can induce apoptosis of vascular smooth muscle cells (82). Thus, diabetes alters vascular smooth muscle function in multiple ways that promote atherosclerotic lesion formation, plaque instability, and clinical events.
Impaired platelet function and increased blood coagulability are also involved in atherogenesis, progression of atheromatous plaque, plaque disruption, and thrombotic occlusion in diabetic patients (83,84,85,86,87,88,89,90).
As the prevalence of diabetes among CHD patients increases (91) and other risk factors such as smoking and hypercholesterolemia are better controlled, diabetes is likely to become the predominant risk factor for atherosclerosis.
Hypertension
Systemic hypertension is well established as a major risk factor for cardiovascular mortality and morbidity in the general population, being more common than cigarette smoking dyslipidemia, and diabetes (92,93). Hypertension is not only a well-established cardiovascular risk factor for the development of atherosclerosis but also appears to increase the risk of atherosclerosis (94,95,96). The INTERHEART study showed that hypertension accounted for 18% risk of a first myocardial infarction (26).
Increased blood pressure may damage the vessel wall directly and serve as a stimulus for inflammation (97). Indeed, impaired fibrinolysis in patients with hypertension may play an important role in the pathogenesis of cardiovascular disease in hypertensive patients (98).
There is increasing evidence that hypertension, through the vasoactive peptides angiotensin and endothelin-1, promotes and accelerates the atherosclerotic process via inflammatory mechanisms. Angiotensin II does decrease NO bioavailability by promoting oxidative stress (99) and may induce intimal inflammation and elicit the production of superoxide anion, a reactive oxygen species, from endothelial cells (100,101,102). It can also increase expression of proinflammatory cytokines and leukocyte adhesion molecules.
Indeed, angiotensin II–induced hypertension accelerates the development of atherosclerosis in ApoE-deficient mice (103). Therapeutic blockade of angiotensin II using angiotensin-converting enzyme inhibitors or angiotensin II receptor blockers in experimental studies and in clinical trials has been shown to correct ED and to retard the progression of atherosclerosis, in part related to the decreased expression of inflammatory mediators and improved endothelial function (104,105,106).
Endothelin-1 is another important mediator of chronic inflammation in the vascular wall. It is capable of inducing NFkB expression and increasing the expression on CD40L. It also mediates vasoconstriction and is expressed at high levels in carotid plaques. Chronic inhibition of endothelin reduces fatty streaks and plaque burden, and restores NO-responsiveness of the endothelium.
Other Risk Factors
Obesity not only predisposes to insulin resistance and diabetes, but also contributes to atherogenic dyslipidemia. The resulting elevation in VLDL from visceral fat can lower HDL cholesterol by augmenting exchange from HDL to VLDL by cholesteryl ester transfer protein. Adipose tissue itself can give rise to cytokines that worsen insulin sensitivity and provide a systemic proinflammatory stimulus (109).
Although not yet truly identified as risk factor, morbid obesity and the resultant metabolic syndrome, a current worldwide epidemic, are setting the stage for type 2 diabetes, its microvascular complications, and acceleration of macrovascular disease. Insulin resistance, hyperglycemia, dyslipidemia, hypertension, and thrombotic disorders as well as adiposity define the metabolic syndrome and contribute to ED and atherosclerosis.
In the metabolic syndrome, LDL particles may have qualitative alterations reducing their size and their vulnerability to oxidation. The low levels of HDL and elevated levels of triglyceride may blunt other endogenous anti-inflammatory mechanisms (110,111).
Nontraditional, emerging risk factors include lipoprotein(a); homocysteine; infectious agents such as Chlamydia pneumoniae, cytomegalovirus, and herpesvirus; and oxidant stress evoked by angiotensin II.
Elevated homocysteine levels appear to injure endothelial cells and stimulate proliferation of vascular smooth muscle cells (SMC) (112).
Infection by cytomegalovirus has been linked to atherosclerosis and arterial restenosis (113). Infection with C. pneumoniae is the most widely studied and may contribute to formation and progression of atherosclerotic lesions (114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129). The obligate intracellular bacterium has been detected in atherosclerotic lesions by immunohistochemistry, polymerase chain reaction, and electron microscopy and has also been cultured from atheromatous plaques (115,116,117,118,119). SMCs and macrophages in the intima have been found to be infected by C. pneumoniae (120,121). If the inflammatory response does not effectively neutralize or remove the offending agents, it can continue indefinitely, resulting in progression (122). Treatment with antibiotics such as azithromycin, however, has not been consistently shown to reduce the number of cardiovascular events of atherosclerosis progression.
Psychiatric manifestations, such as depression, hostile behaviors, and anger expression, can affect clinical outcome and may play a role in atherosclerotic progression (130,131,132,133,134).
Family history of CHD is a major risk factor, particularly when it involves an immediate relative with premature coronary disease (135).
Exercise is considered an antiatherogenic factor, thus lack of activity and a sedentary lifestyle may be permissive for atheroma formation (136).
Pathogenesis
It is now clear that systemic and local inflammatory events mediate plaque formation, progression, and degeneration: Atherosclerosis is a chronic inflammatory disease (33,122,137,138,139,140,141,142,143). No longer regarded as a bland, simple, and degenerative process, plaque evolution is best illustrated as a continuous tug of war between proinflammatory and anti-inflammatory cellular and molecular pathways.
Animal models have enabled a better understanding of the pathways involved. Targeted disruption of the apolipoprotein E (Apo E) or LDL receptor genes or overexpression of the human apolipoprotein B (Apo B) gene in mice resulted in marked increases in plasma cholesterol levels and development of advanced atherosclerotic lesions (144,145). While providing great insights into pathogenesis of atherosclerosis, the animal models currently available have important limitations in their applicability to the human disease.
In virtually all animal models, atherosclerosis is driven by extreme elevations in circulating cholesterol levels over a time scale of weeks to months. In contrast, atherosclerosis in humans is developed over decades. In autopsy studies, men and women, aged 15 to 34 years, had aortic fatty streaks with the more advanced lesions occurring with increasing age (146,147,148), and inflammation was at the center of it all. In fact, there is increasing evidence that the classically imagined “response-to-injury” is truly an inflammatory reaction (141,142).
Histologic Classification of Atherosclerotic Plaque
Atherosclerotic disease has been classified as a six-stage stepwise process of cross-sectional morphologic and histologic change (148,149,150) (Fig. 7-2). A normal artery evolves into types I and II initial lesions of atherosclerosis. These lesions have a characteristic architecture of fatty streaks and appear even in childhood at injury-prone areas such as bifurcations (151). The histologic features of atherosclerotic stages are
shown in Table 7-2; a host of complex interactions occur in atherogenesis.
shown in Table 7-2; a host of complex interactions occur in atherogenesis.
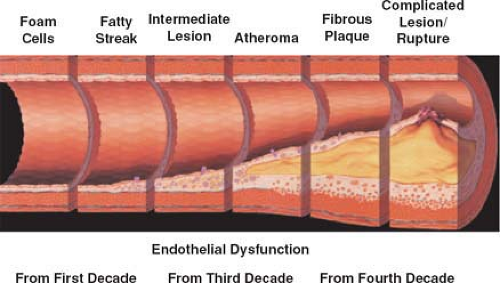 Figure 7-2 Evolution of atherosclerosis from earliest fatty streaks to plaque rupture and myocardial infarction. Adapted from Pepine CJ. Am J Cardiol. 1998. |
Fatty streaks are considered the first phase in atherosclerotic plaque development. Histologically, it presents as focal thickening of the intima with an increase in leukocytes, SMCs, and extracellular matrix (152). The fatty streaks also consist of macrophages with a variable number of T lymphocytes and lipid accumulation. Apoptosis of SMC within the fatty streak is associated with further macrophage infiltration and cytoplasmic remnants that contribute to the transformation of fatty streaks into atherosclerotic plaques (153).
Subsequently, fatty streaks progress into fibrous plaques via accumulation of connective tissue with an increased number of smooth muscle cells and lipid-laden macrophages. More advanced lesions often contain a necrotic lipid-rich core and receive vascular supply from both the luminal and medial sides.
A spectrum of possible advanced lesions can result, ranging from the chronic stable type IV atheroma of adulthood to the complicated type VI plaque of acute coronary syndromes. Furthermore, long-standing repetitive inflammatory injury can be the primary factor stimulating the transformation of stable mature type IV lesions into type V fibroatheromas prone to symptomatic luminal stenosis or degenerated type VI plaques vulnerable to acute rupture, hemorrhage, ulceration, thrombosis, or embolization (137,154).
Table 7.2 Criteria for American Heart Association Lesion Classification System and Correspondence with Classification of Gross Arterial Specimens | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||
Initiation of Atherosclerosis and Inflammation
Endothelial Dysfunction
The endothelium is not an inert, single-cell lining covering the intima of arteries, but in fact plays an essential role in regulating vascular tone and structure. It also functions as a selective permeability barrier between blood and tissues and can produce various molecules essential for regulation of thrombosis, inflammation, and vascular remodeling. In atherogenesis, ED is the earliest measurable functional abnormality of the vessel wall. It is a consequence of the harmful effects of the risk factors of atherosclerosis on the vessel wall.
Under healthy circumstances, the endothelial monolayer resists firm adhesion of platelets and leukocytes and maintains a balance of profibrinolytic and prothrombotic activity (33). Removal of the endothelium results in platelet adhesion and in a burst of SMC migration and proliferation, the latter subsiding when the endothelium regenerates (155).
Common factors predisposing to atherosclerosis such as hypercholesterolemia and diabetes are associated with ED, which might be the initial step in atherogenesis. Both local and systemic factors such as mechanical denudation
(angioplasty), advanced glycation end products, infectious agents, uremia, cigarette toxins, oxidative stress, genetic variability, radiation injury, homocysteinemia, chronic autoimmune conditions, and shear stress are associated with ED (156,157,158,159,160,161,162,163).
(angioplasty), advanced glycation end products, infectious agents, uremia, cigarette toxins, oxidative stress, genetic variability, radiation injury, homocysteinemia, chronic autoimmune conditions, and shear stress are associated with ED (156,157,158,159,160,161,162,163).
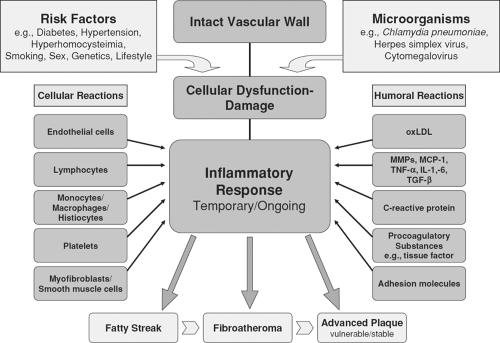 Figure 7-3 Schematic construction of the interrelationship between cellular and humoral factors involved in the inflammatory response of atherogenesis. |
Endothelial cells in regions of arterial branching with turbulent blood flow have polygonal shape and no particular direction, while in areas with uniform flow, endothelial cells are ellipsoid in shape and aligned in the direction of flow. An absence of laminar flow may reduce local production of endothelium-derived nitric oxide (142).
ED is also associated with increased oxidative stress, an important promoter of the inflammatory processes. Oxidative stress is a harmful condition that occurs when there is an excess of free radicals, a decrease in antioxidant levels, or both.
NO is a potent oxidant and chemical mediator with multiple anti-atherogenic properties, including vasodilatory and anti-inflammatory properties. Its production is impaired in ED.
Experimental studies have demonstrated that leukocyte adhesion and infiltration into the intima, an essential step in atherosclerotic lesion formation, is regulated by leukocyte adhesion molecules and chemokines (164,165,166).
Pharmacologic inhibition of endothelium-derived NO production results in marked increase in endothelial adhesiveness for monocytes (167,168), an effect attenuated by dietary L-arginine, the substrate of endothelial NO synthase (eNOS).
At the molecular level, inhibition of eNOS results in increased expression of leukocyte adhesion molecules and critical chemokines, which are thought to be responsible for the migration of monocytes into the intima at sites of atherosclerotic lesion formation (169).
In contrast, NO synthase gene therapy dramatically reduces hypercholesterolemia-induced leukocyte adhesion molecule expression and inhibits monocyte infiltration into the arterial wall in a hypercholesterolemic rabbit model (170). Accelerated atherosclerotic lesion formation in the
aorta has been demonstrated in eNOS knockout mice (171). Thus, NO serves as a protective moiety. ED with impaired NO production makes the affected intima more vulnerable.
aorta has been demonstrated in eNOS knockout mice (171). Thus, NO serves as a protective moiety. ED with impaired NO production makes the affected intima more vulnerable.
Following a variety of injuries, including shear stress, the endothelial cells respond with an expression of adhesion molecules, increase their permeability, undergo further endothelial cell proliferation, and initiate thrombosis (137).
When the physiologic properties of endothelial cells are altered, several endothelium-derived adhesion molecules are expressed, leading to attachment and accumulation of monocytes, macrophages, T lymphocytes, and platelets on the arterial wall.
Both endothelial and attached cells become activated and release proinflammatory cytokines, membrane receptors, and enzymes. By-products of this cell activation include expression of interleukins (IL) 1, 2, 6, 7, 8, and 18; tumor necrosis factor-α(TNF-α); interferon-γ (IFN-γ); monocyte chemotactic protein (MCP-1); MCP-4; CD40 ligand (CD40L); parathyroid hormone-related protein (PTHrP); osteopontin; cyclo-oxygenase-2 (COX-2); and MMPs (172,173,174




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
