Introduction
Monogenic diseases are caused by single genetic defects, follow Mendelian inheritance, and are classified as autosomal dominant, autosomal recessive or X-linked. Over 5000 monogenic diseases have been identified, and more than 1000 genes responsible for these disorders are known. The prevalence of monogenic diseases ranges from 1 in 1000 individuals in the most common forms to 1 in 200 000 in the rarest. The identification and study of the genetic substrates of these diseases are important not only because they contribute to the development of diagnostic tests for affected individuals but also because they promote understanding of the function of different proteins in humans. Consequently, genetic testing is currently applied not only for diagnostic evaluation, but also as a research tool for the study of pathophysiology of inherited arrhythmia syndromes. In cardiology, there are two major clusters of monogenic disorders: the cardiomyopathies due to alterations in sarcomeric and in cytoskeletal proteins, and the arrhythmogenic diseases that are caused by mutations in ion channels and ion channel-controlling proteins, e.g. the congenital long QT syndromes (LQTS), the Brugada syndrome, catecholaminergic polymorphic ventricular tachycardia (CPVT), Andersen–Tawil syndrome, conduction system disease, and atrial fibrillation (AF).
What we have learned from the genetics of monogenic arrhythmia syndromes
The identification of genes responsible for the inherited arrhythmia syndromes has contributed greatly to our understanding of the substrate for arrhythmia development. More importantly, it has provided practical information when managing affected patients with these disorders. The identification of a mutation allows us to suspect the diagnosis independent of the clinical features of the syndrome. When screening family members of a genotyped proband, an unexpectedly large number of carriers is identified among relatives initially considered unaffected based on clinical phenotyping. For example, up to 32% of carriers of LQTS-related mutations can have a normal QTc.1 As silent carriers have a 50% probability of transmitting LQTS to their offspring and they are more likely to develop cardiac arrhythmias compared to an age-matched population,2 knowing the genotype status of an individual has practical implications. In addition, silent carriers of a LQTS-related genetic defect still have a 20% risk of becoming symptomatic.2 Thus, the most immediate contribution of genetic information to clinical practice is that of providing a novel and useful parameter for risk stratification that can be used alone or in combination with other clinical variables. Currently, only in the congenital LQTS do we have data on hundreds of genotyped patients with long clinical follow-up but emerging data for other diseases such as CPVT are becoming available. As progress is made in our understanding of the links between DNA defects and clinical phenotype, genotyping patients with inherited arrhythmia disorders is even more likely to contribute to the clinical management of patients.
Complexity beyond monogenic arrhythmia disorders
Since the initial discovery of mutations in KCNQ1, KCNH2, and SCN5A cardiac ion channel genes by Keating and colleagues over a decade ago,3–5 hundreds of mutations have been identified in various ion channel subunits and in proteins important for the proper functioning of cardiac ion channels. Although it was initially assumed that all carriers of pathogenic mutations would manifest the corresponding phenotype, it soon became apparent that the clinical consequences of genetic defects are far more variable than expected. Some of this variability is related to incomplete penetrance and variable expressivity. The penetrance of a monogenic disease is defined as the percentage of individuals with a mutant allele who develop the phenotype of the related disease and it can vary from 10% to 100%. The expressivity of a disease is defined as the different phenotypical manifestations that can be observed among carriers of the same genetic defect. Consequently, the combination of variable penetrance and expressivity dictates that carriers of a DNA mutation may manifest either no clinical phenotype or phenotypes that are not the classic “textbook” type. Thus, genotyping patients affected by “simple” monogenic diseases is more complicated than initially envisioned.
Another important concept when considering monogenic diseases is genetic heterogeneity. Analogous to the genetics of inherited cardiomyopathies, in arrhythmia disorders the “same” phenotype is associated with multiple genetic defects. At present, nine genes are known to cause the phenotype of the congenital LQTS and explain 60–75% of the clinically diagnosed cases. Similarly, two genes have been associated with CPVT and they account for half of the patients with the phenotype.6 Two genes are known to cause the Brugada syndrome, but several other genes are likely to be implicated because so far only 20% of clinical diagnoses result in positive results at genetic testing.7 Perhaps an obvious question that stems from these considerations is whether we should refer to all genetic variants with the same name, such as LQTS. Should all individuals with the same ECG marker, e.g., prolonged QT interval, ST segment elevation in the right precordial leads, adrenergically driven polymorphic ventricular tachycardia (VT), be grouped under the same diagnosis, when we know that each genetic variant has a distinguishing clinical phenotype? This may be more than semantics as there are emerging data that the genetic substrate is a major determinant of prognosis in patients harboring different genetic mutations.2
In the future we may consider each genetic variant as a separate disease based on the specific defect as this would avoid the difficulty of trying to fit the complex and overlapping phenotypes into clinical categories. This would not only simplify the categorization of overlapping syndromes but also the clinical profiles: Brugada syndrome with conduction defects, Brugada syndrome with atrial arrhythmias, conduction defects with sinus abnormalities, Brugada syndrome with prolonged QT interval, etc. All these conditions could be grouped in the category of sodium channel disease and its full spectrum of abnormalities. This is complicated even further as it is now established that mutations in one gene may lead not only to variable phenotypes within the same disease, but also to profoundly different diseases. The most emblematic example of this diversity is represented by mutations in the lamin A/C gene, which are known to cause at least eight phenotypes (allelic diseases) as different as dilated cardiomyopathy, Emery–Dre-ifuss muscular dystrophy, familial partial lipodystrophy, Charcot–Marie–Tooth disease, mandibuloacral dysplasia, Hutchinson–Gilford progeria, lipoatrophy with diabetes, and hepatic steatosis hypertrophic cardiomyopathy. In inherited arrhythmia disorders, three diseases have been associated with mutations of the cardiac sodium channel gene (LQTS, Brugada syndrome, and progressive conduction disturbances)4,8 and three diseases (LQTS, short QT syndrome and familial AF) have been associated with mutations in the KCNQ1 gene encoding for the alpha-subunit of the potassium channel conducting the slow component of the delayed rectifier (I Ks).5,9,10 It is therefore obvious that the identification of a mutation in a given gene cannot establish the diagnosis of a single disease and the identification of a mutation in an individual with a known disease is not enough to predict the phenotype of that individual.
We will now discuss individual inherited monogenic arrhythmia disorders.
Atrial fibrillation (AF), the most common cardiac arrhythmia in clinical practice, affects approximately 2% of the United States population and results in substantial morbidity and mortality.11 The increasing prevalence of AF parallels the increasing age of the general population.12 AF is also the most common arrhythmia requiring drug therapy. The limited success of current interventions for AF is partially the result of heterogeneity of the underlying electrical substrate and incomplete understanding of fundamental mechanisms in disease pathogenesis. Although some of the risk factors associated with AF have been identified, relatively little is known about the underlying molecular events leading to the arrhythmia.13 One approach to unraveling the molecular pathogenesis of AF is through the identification of genes responsible for familial forms of the disease. Evidence for the heritability of AF has come from several sources including the study of AF kindreds who exhibit the arrhythmia as a primary disease, the analysis of AF presenting in the setting of another familial cardiac disease, and the analysis of common population genetic variants, i.e. DNA polymorphisms, that may predispose to AF.
Monogenic familial AF was first reported in 194314 and while it may be uncommon, there has been no attempt to determine the prevalence of familial AF. Heritability of AF is further suggested by two recent population-based studies demonstrating that AF in first-degree relatives was associated with an increased risk of developing AF.12,15 One study showed that 5% of patients with AF and up to 15% of individuals with lone AF may have a familial form of the disease.16,17
A gene locus for AF was first reported in 1997, based on genetic mapping in three Spanish families.18 However, the gene responsible for AF in these kindreds has not yet been identified, but resides within a relatively large chromosomal region spanning 14 centiMorgans (cM). In addition, three other loci on chromosomes 6q14–16,19 5p1320 and 10p11-q2121 have been reported, with no genes yet identified. In 2002, a novel AF locus on chromosome 5p15 was identified by genome-wide linkage analysis of a four-generation kindred.22 Importantly, this study established an abnormally prolonged P-wave (>155ms) determined by signal-averaged ECG analysis as an “endophenotype” that improved statistical power of the linkage study in this family. Collectively, these reports support the idea that familial AF is a genetically heterogeneous disease much like many other inherited arrhythmia syndromes.16
Recently, the first genes for AF have been identified, providing a link between ion channelopathies and the arrhythmia. In a four-generation Chinese family in which LQTS and early-onset AF co-segregate, a mutation (S140G) in KCNQ1 gene on chromosome 11p15.5 has been reported.10 Functional analysis of the S140G mutant revealed a gain-of-function effect in KCNQ1/KCNE1 and KCNQ1/KCNE2 currents, which contrasts with the dominant negative or loss-of-function effects of KCNQ1 mutations previously associated with LQTS. More recently, the same group established a link between KCNE2 and AF by identification of a mutation in two families with AF.23 The mutation R27C also caused a gain-of-function when co-expressed with KCNQ1 but had no effect when expressed with KCNH2. Recently, a truncating mutation in KCNA5 encoding a voltage-gated potassium channel (K V 1.5) underlying the ultra-rapid delayed rectifier current (I Kur) was associated with familial AF.24 These studies firmly establish the role of potassium channels in the pathogenesis of some forms of familial AF, but resequencing studies in large cohorts suggest that mutations in these channels are a rare cause of the arrhythmia.25
AF associated with other monogenic diseases
Studies of other cardiac monogenic disorders have also provided evidence for the genetic contribution to the etiology of AF. These include diseases such as hypertrophic cardiomyopathy, skeletal myopathies, familial amyloidosis, and atrial myopathies. It is likely that AF in these cases is related at least in part to non-specific structural changes in the atria caused by the underlying cardiac pathology, but AF is uniquely prominent in some kindreds with each of these conditions.26 AF can also present in other ion channelopathies like congenital LQTS type 4,27 Brugada syndrome28 and short QT syndrome.29 The high incidence of atrial arrhythmias in patients with short QT syndrome and gain of function mutations in IKs30 point to an important role for shortening of the action potential in the development of AF. Sodium channel gene (SCN5A) defects have also been associated with a syndrome of early-onset dilated cardiomyopathy and AF.31 Moreover, mutations in the gene for the nuclear membrane protein lamin A/C (LMNA) have pleiotropic non-cardiac and cardiac manifestations including dilated cardiomyopathy, AF, and conduction system disease.32 Collectively, these studies attest to the heterogeneous nature of AF and strongly suggest that defects in many more genes remain to be identified.
AF associated with cardiac or systemic diseases
Most patients with AF have one or more identifiable risk factors, but many or even most patients with these same risk factors do not develop AF. Thus, it is likely that genetic determinants favor AF in some individuals with identifiable risk factors. Studies comparing cases of non-familial AF to age- and gender-matched controls (association studies) have provided insight into the genetic basis of “acquired” AF. One study evaluated a polymorphism in KCNE1 and AF and identified an association with the 38G allele. While the 38G allele appears to reduce IKs,33 mice lacking KCNE1 are prone to AF due to an unexpected increase in IKs,34 suggesting that the consequences of ion channel mutations are not always straightforward. In addition, common DNA polymorphisms in GNB3,35 KCNE5,36 and SCN5A have all been associated with AF.37 Over the last several years increasing evidence has arisen that renin-angiotensin-aldosterone (RAAS) activation may be an important risk factor for the development of AF. Retrospective analyses suggest that angiotensin-converting enzyme (ACE) inhibitor therapy is associated with a lower incidence of AF and a placebo-controlled trial found a similar beneficial effect of adding the angiotensin receptor blocker (ARB) irbesartan to amiodarone.38 Additionally, a case–control study of 250 Taiwanese subjects with AF and 250 controls identified polymorphisms in this pathway as risk factors for AF.39 Added support for the role of RAAS activation in the pathophysiology of AF comes from a recent study which demonstrated a pharmacogenetic interaction between the ACE I/D polymorphism and efficacy of antiar-rhythmic drug therapy in patients with lone AF.40
The above studies in aging patients with non-familial AF in the presence of underlying heart disease suggest some form of heritable contribution to the pathogenesis of the more common forms of AF. These data are promising and may help clarify why some people develop AF under specific circumstances while others may not.
Uncovering common genes for AF
A logical consequence of the availability of comprehensive genomic maps is the advent of high-density genome-wide searches for modest gene effects using large-scale testing of single nucleotide polymorphisms (SNPs). Such an approach has been suggested to tackle complex human diseases such as AF. Early proponents suggested the study of coding or promoting variants with potential functional significance. Collins et al subsequently proposed that non-coding or evenly spaced SNPs with high density could be used to track disease loci through linkage disequilibrium.41 The availability of high-density mapping of marker SNPs and assessment of genomic structure, together with emerging information on functional pathways, have begun to provide powerful means of identifying genetic susceptibilities to AF. In the first genome-wide association study of AF, a novel locus on chromosome 4q25 has recently been identified which confers a 1.6–2.0 (95% confidence interval)-fold increased risk of the arrhythmia across multiple different populations.42
While there is significant overlap among the risk factors for AF and atrial flutter, in general these arrhythmias exhibit quite distinct biology and electrophysiology.13 Atrial flutter is commonly associated with the later stages of a broad range of congenital heart disease (CHD). There is extensive heterogeneity particularly in the most profound structural disorders, but increasing evidence of a heritable component to many forms of CHD is accumulating. The association of CHD with atrial flutter has been attributed to abnormal atrial patterning, which may range from the extreme to the subtle, often with no other obvious manifestations. Interestingly, atrial flutter was found to be more strongly associated with the locus on chromosome 4q25 than AF, at least suggesting that there may be common factors underlying the clinical presentation of some forms of these two arrhythmias.42 Given the intrinsic biases of genome-wide association studies, these links might be far downstream of the primary causation, but nevertheless may have substantial effects on a population basis.
Classic Mendelian genetics have revealed several distinctive mechanisms for sinus nodal disease. Mutations in the cardiac transcription factors Nkx 2.5 and Tbx 2.5 both cause defects in the differentiation and maintenance of specialized conduction tissues (particularly those of the AV node) and each causes subtle abnormalities of sinus impulse generation.8,43–45 Similarly, several primary arrhythmic syndromes including some forms of the LQTS also result in perturbations of sinus rhythm.46 While in many instances potential mechanisms have been framed in terms of effects on transmembrane ion currents, data from genetic models of SCN5A and AnkB disease suggest that subtle trophic effects on myocardial structures may also play a role.47,48 Genetic models may also inform human studies at an earlier stage. Murine null alleles of the pacemaker channels HCN2 and HCN4 first directly implicated these channels in normal sinus rhythm, and subsequent human genetic studies have identified mutations in the HCN4 gene in sinoatrial disease.49–52
Table 44.2 Genetic arrhythmia syndromes associated with ventricular arrhythmias
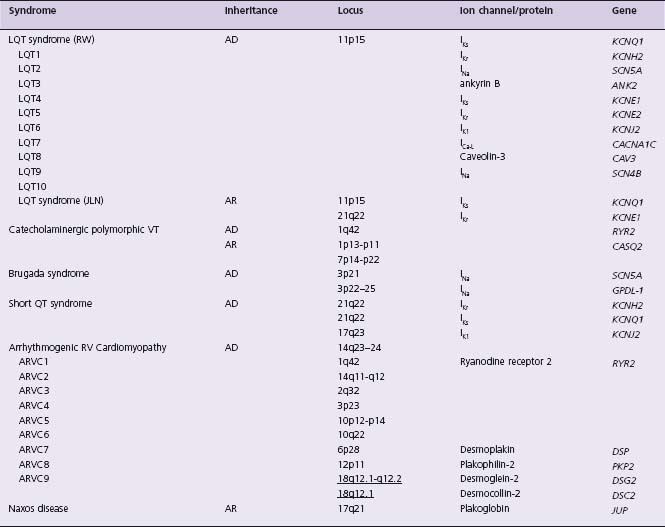
JLN, Jervell–Lange-Nielsen; LQT, long QT; RV, right ventricular; RW, Romano–Ward; VT, ventricular tachycardia. See Table 44.1 for remaining abbreviations.
Table 44.1 Atrial arrhythmias due to monogenic disorders
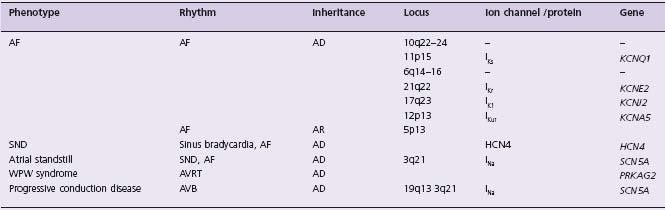
AD, autosomal dominant; AF, atrial fibrillation; AR, autosomal recessive; AVRT, atrioventricular re-entrant tachycardia; AVB, atrioventricular block; SND, sinus node dysfunction; WPW, Wolff–Parkinson–White.
Virtually every known myocardial disease has been associated with some form of atrial arrhythmia. However, there are some rare associations which appear to be considerably more specific. A number of metabolic disorders, including mitochondrial diseases, glycogen or other storage disorders, are strongly associated with prominent evidence of AV conduction disease, often with overt ventricular pre-excitation as well as frequent macro re-entrant and other atrial arrhythmias. For example, dominant mutations in the AMP-activated protein kinase gamma subunit (PRKAG2) have been shown to result in massive myocardial thickening, AV conduction system disease and ventricular pre-excitation.53 These families had previously been included under the rubric of hypertrophic cardiomyopathy on the basis of their inheritance patterns, adult-onset and echocar-diographic features. AF and atrial flutter are common, but high-grade AV block is the dominant clinical arrhythmia in these kindreds. Clinical studies suggest that in many cases asymptomatic individuals are maximally pre-excited at rest, and therefore probably dependent on accessory AV connections from an early age.54 These data defined a novel subset of “hypertrophic cardiomyopathy” illustrating the potential utility of molecular nosology for cardiac disease, but also offer some unique insights into the mechanisms of normal AV electrical development.53
Congenital long QT syndromes
The congenital LQTS are a group of genetic disorders that affect cardiac ion channels, characterized by prolongation of the QT interval, and risk for the life-threatening ventricular tachycardia torsades de pointes. Shortly after the autosomal recessive syndrome of congenital deafness, prolongation of the QT interval, and sudden death was described by Anton Jervell and Fred Lange-Nielsen in 1957,55 Romano and Ward each independently described an “autosomal dominant” form without congenital deafness.56,57 In the late 1990s the first five LQTS genes were identified, all of which encode ion channel subunits that underlie the cardiac action potential.3–5,58 The most commonly affected genes, KCNQ1 and KCNH2 (underlying LQT1 and LQT2 respectively), encode proteins that form the alpha-subunits of two major repolarizing potassium currents, IKs and IKr. Two other LQTS genes encode for the corresponding beta-subunits (KCNE1 and KCNE2 underlying LQT5 and LQT6, respectively). The other major LQTS gene, SCN5A (underlying LQT3), encodes the alpha-sub-unit of the cardiac sodium channel. It is now accepted that patients with “Romano–Ward syndrome” carry a single mutation, while homozygous mutations (or compound heterozygotes) of KCNQ1 or KCNE1 cause Jervell–Lange-Nielsen (JLN) syndrome.59,60 The extracardiac finding of congenital deafness requires the presence of two mutant alleles, and results from lack of functioning IKs in the inner ear.61 JLN syndrome patients are also highly susceptible to arrhythmias; thus arrhythmia risk seems partly dependent on “gene dosage.”62 These five classic forms of congenital LQTS result from mutations which reduce outward currents (I Ks or I Kr) or enhance inward current (I Na), thereby prolonging action potential duration and, in turn, prolonging the QT interval.
Additional ion channel mutations have been associated with rare arrhythmia syndromes (Andersen–Tawil syndrome, KCNJ2, and Timothy syndrome, CACNA1C) which may include QT prolongation, as well as significant extracardiac phenotypes.63,64 Andersen–Tawil syndrome patients do not uniformly display prolonged QT intervals, and due to clinical features that differ from LQTS, this syndrome is better termed ATS1 rather than LQT7.65 The previously termed LQT4 has been linked to mutations in ANK2, encoding the structural protein ankyrin-B, which when mutated results in altered localization and expression of ion channels.27 Patients with ANK2 mutations do not uniformly have prolonged QT intervals, and it has been suggested that LQT4 be renamed “sick sinus syndrome associated with bradycardia” or “ankyrin-B syndrome.”66
The incidence of congenital LQTS has been estimated at 1 in 5000, and the incidence of JLN syndrome estimated as 1 in 500 congenitally deaf individuals. Without treatment, 13% of LQTS patients will suffer cardiac arrest or sudden death prior to 40 years of age; when syncopal events are included, 36% will have symptoms by age 40.2 The JLN syndrome is more severe, with 90% experiencing syncope, cardiac arrest or sudden death by age 18, and mortality exceeding 25% even with therapy.62 Important clinical differences among affected patients depending on the underlying affected gene have been observed–so-called genotype-phenotype correlation. As the majority (>90%) of genotyped LQTS patients have LQT1, LQT2 or LQT3,67 most of the differences are observed among these genotypes and include different ECG T-wave patterns,68 clinical course,69 triggers of cardiac events,70 response to sympathetic stimulation,71,72 and effectiveness and limitations of beta-blocker therapy.73
Pharmacologic therapy for LQTS
Despite the genotype-specific aspects mentioned above, evidence-based therapeutic recommendations, for the most part, disregard the underlying genotype.74 This is practical, as approximately one-third of patients with an unambiguous clinical diagnosis of LQTS do not have mutations in any of the known LQTS genes. Beta-blockers and lifestyle modifications, including restriction from vigorous exercise and avoidance of drugs which prolong the QT interval, are recommended for all patients with a clinical diagnosis of LQTS73 (Class I, Level B). While there are no placebo-controlled clinical trials of beta-blockers in LQTS, the data from retrospective comparisons of cohorts with and without therapy, event rates before and after beta-blocker therapy, and data from the International Registry provide convincing evidence (60% reduction in risk for cardiac events, and LQTS-related death).73,75,76 Aside from beta-blockers no pharmacologic intervention has been supported by enough data to warrant evidence-based recommendations. Sodium channel blockers seem a logical choice for patients with LQT3, and mexiletine has been shown to shorten the QT interval both in in vitro models of LQT3 and in a small cohort of LQT3 patients.77,78 Concerns about the safety of flecainide were raised when intravenous flecainide elicited a Brugada syndrome phenotype in six out of 13 LQT3 patients.79 However, in eight LQT3 patients with a specific SCN5A mutation, oral flecainide shortened the QT and was safely tolerated chronically.80 Elevating serum potassium, both acutely81 and chronically with oral potassium and spironolactone, shortens the QT interval in LQT2.82 Despite these potentially beneficial effects on repolarization, a reduction in cardiac events has yet to be demonstrated with any drug other than beta-blockers.
Non-pharmacologic therapy for LQTS
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


