Introduction
The primary purpose of this chapter is to review the evidence regarding plasma lipid-altering medications, their mechanisms of action, dosages and dosing schedules, effects on lipid and lipoprotein variables, adverse effects, and clinical uses. The 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitor agents (statins), which are highly effective LDL-C lowering agents, are reviewed, as are niacin, bile acid-sequestering agents (resins), fibrates, and ezetimibe. The evidence for cholesterol lowering in such subgroups as the elderly, women, diabetic patients, and those with small-dense LDL particles is also summarized.
Major trials have clearly demonstrated that decreases in low-density lipoprotein cholesterol (LDL-C) are associated with reductions in total mortality,1–3 coronary heart disease (CHD) mortality,1–3 fatal and non-fatal CHD as well as strokes.1–4 Other major trials have also shown that lowering LDL-C can retard the progression of coronary artery atherosclerosis5 and carotid atherosclerosis and may even cause their regression, as well as slowing the progression and occlusion of atherosclerosis in saphenous vein bypass grafts.6 In both primary2 and secondary1,3 CHD prevention settings, decreases in total and cause-specific mortality have been demonstrated, and these benefits have been shown in subjects with elevated,1,2 average,4 and normal LDL-C levels. A brief review of the costs/1% LDL-C lowering/year and cost effectiveness concludes this chapter.
The National Cholesterol Education Program (NCEP) has been instrumental in developing and promulgating guidelines for initiating LDL-C lowering. These guidelines, developed on the basis of the patient’s established baseline LDL-C and presence or absence of CHD or its risk factors, recommend treatment goals to attain desired levels of plasma LDL-C. To date, the NCEP has issued three Adult Treatment Panel (ATP) reports. ATP I emphasized primary prevention of CHD in persons with high (>160mg/dL or 4.15mmol/L) or borderlinehigh LDL levels (130–159mg/dL or 3.18–4.15mmol/L) and >2 risk factors for development of CHD. In ATP II, persons with established CHD were targeted for intensive lipid-lowering therapy.
ATP III, disseminated in 2001, identifies elevated LDL-C as the primary target of cholesterol-lowering therapy and maintains attention on intensive treatment of patients with CHD.7 It expands the indications for intensive therapy to lower levels of cholesterol in clinical practice. A major new feature is that intensive LDL-C lowering treatment is a primary prevention measure for persons with multiple risk factors for developing CHD, as identified by the estimated 10-year CHD risk score developed from the Framingham data. ATP III sets the optimal LDL-C level as <100mg/dL (2.60mmol/L) and defines low HDL-C as <40mg/dL (1.04mmol/L) (previous cutpoint was <35mg/dL (0.91mmol/L)).7
In 2004, recommendations for modifications to footnote the ATP III LDL-C treatment algorithm were issued.8 Based upon the results of five major clinical outcome trials completed since the ATP III guidelines were published, a therapeutic option of an LDL-C goal of <70 mg/dL (1.82 mmol/L) for those at very high risk was considered a reasonable clinical strategy. This more aggressive goal was also extended to those at high risk who have baseline LDL-C < 100 mg/dL (2.60 mmol/L). In addition, it was recommended that those at high risk who also have high triglycerides, or low HDL-C levels, be considered for combination therapy of nicotinic acid or a fibrate along with an LDL-C lowering agent. When those at high risk, or moderately high risk, are treated with LDL-lowering therapy, it was recommended that the intensity of treatment be sufficient to achieve at least a 30–40% reduction in LDL levels.8
ATP III also recommends that persons with the metabolic syndrome – characterized by abdominal obesity, elevated blood pressure, insulin resistance, and atherogenic dyslipidemia–elevated triglycerides, small LDL particles, and low HDL-C should be targeted for intensive therapeutic lifestyle modifications. Atherogenic dyslipidemia should be treated with lipid-altering agents.7
It has recently been demonstrated by the INTERHEART study that the Apo B to Apo A1 ratio will most completely capture the risk associated with dyslipidemia.9 The role of triglycerides as an independent risk factor for the development of atherosclerosis remains controversial. The risks and therapeutic benefits of treatment have been recently reviewed.12
BOX 12.1 New features of adult treatment panel III
- Focus on multiple risk factors
- Raises persons with diabetes without CHD, most of whom display multiple risk factors, to the risk level of CHD risk equivalent
- Uses Framingham projections of 10 year absolute CHD risk (that is, the percent probability of having a CHD event in 10 years) to identify certain patients with multiple (2+) risk factors for more intensive treatment
- Identifies persons with multiple metabolic risk factors (metabolic syndrome) as candidates for intensified therapeutic lifestyle changes
- Modifications of lipid and lipoprotein classification
- Identifies LDL-C <100mg/dL (2.60mmol/L) as optimal
- Raises categorical low HDL-C from < 35 (0.91) to <40mg/dL (1.04mmol/L)
- Lowers the triglyceride classification cutpoints to give more attention to moderate elevations
Adapted from Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III)7
BOX 12.2 ATP III classification of LDL, total and HDL cholesterol
LDL cholesterol:
<100mg/dL (2.60mmol/L) optimal
Near optimal/above optimal
Borderline high
High
≥190mg/dL (4.94mmol/L), very high
Total cholesterol:
<200mg/dL (5.19mmol/L), desirable
Borderline high
≥240mg/dL (6.23mmol/L), high
HDL cholesterol:
<40mg/dL (1.04mmol/L), low
≥60mg/dL (1.56mmol/L), high
Adapted from the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III)7
Boxes 12.1–12.5 summarize the major new recommendations of ATP III and classifications of cholesterol levels.7
Use of individual lipid-altering agents
In this short,evidence-based overview, we focus on documented activities of known lipid-and lipoprotein-altering drugs on lipid and lipoprotein variables, and their related adverse effects. We identify those issues that remain more speculative as such. The interested reader is referred to the excellent and more complete reviews by Lousberg et al,10 as well as the ATP III guidelines.7
BOX 12.3 Major risk factors (exclusive of LDL cholesterol) that modify LDL goals*
- Cigarette smoking
- Hypertension (blood pressure >140/90mmHg or on antihypertensive medication)
- Low HDL cholesterol (<40mg/dL)†
- Family history of premature CHD (CHD in male first-degree relative < 55 years; CHD in female first-degree relative <65 years)
- Age (men >45 years; women >55 years)
*Diabetes is regarded as a coronary heart disease (CHD) risk equivalent.
†HDL cholesterol >60mg/dL (1.56mmol/L) counts as a “negative” risk factor; its presence removes 1 risk factor from the total count.
Adapted from Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III)7
BOX 12.4 Three categories of risk that modify LDL cholesterol goals
| Risk category | LDL goal (mg/dL) |
| CHD and CHD risk equivalents | <100 (2.60mmol/L) |
| Multiple (2+) risk factors)* | <130 (3.38 mmol/L) |
| 0–1 risk factor | <160 (4.16mmol/L) |
*Risk factors that modify the low density lipoprotein (LDL) goal are listed in BOX 12.3. CHD indicates coronary heart disease.
Adapted from Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III)7
BOX 12.5 Recommendations for modifications to footnote the ATP III treatment algorithm for LDL-C
- Therapeutic lifestyle changes (TLC) remain an essential modality in clinical management. TLC has the potential to reduce cardiovascular risk through several mechanisms beyond LDL lowering.
- In high-risk persons, the recommended LDL-C goal is <100mg/dL (2.60mmol/L)
An LDL-C goal of <70mg/dL ((1.82mmol/L) is a therapeutic option on the basis of available clinical trial evidence, especially for patients at very high risk (e.g. continued smoking).
– If LDL-C is >100mg/dL, an LDL-lowering drug is indicated simultaneously with lifestyle changes.
– If baseline LDL-C is <100mg/dL, institution of an LDL-lowering drug to achieve an LDL-C level < 70 mg/dL is a therapeutic option on the basis of available clinical trial evidence.
– If a high-risk person has high triglycerides or low HDL-C, consideration can be given to combining a fibrate or nicotinic acid with an LDL-lowering drug. When triglycerides are >200mg/dL (2.26mmol/L), non-HDL-C is a secondary target of therapy, with a goal 30mg/dL (0.34mmol/L) higher than the identified LDL-C goal. The risks and therapeutic benefits of treatment associated with elevated levels of triglycerides have recently been reviewed.12
- For moderately high-risk persons (2+ risk factors and 10-year risk 10–20%), the recommended LDL-C goal is <130mg/dL (3.38mmol/L); an LDL-C goal <100mg/dL (2.60 mmol/L) is a therapeutic option on the basis of available clinical trial evidence. When LDL-C level is 100–129mg/dL (2.60–3.38mmol/L) at baseline or on lifestyle therapy, initiation of an LDL-lowering drug to achieve an LDL-C level <100mg/dL(2.60mmol/L) is a therapeutic option on the basis of available clinical trial evidence.
New evidence suggests that the Apo B to Apo A1 ratio will most completely capture the risk associated with dyslipidemia.9
- Any person at high risk or moderately high risk who has lifestyle-related risk factors (e.g. obesity, physical inactivity, elevated triglyceride, low HDL-C or metabolic syndrome) is a candidate for TLC to modify these risk factors regardless of LDL-C level.
- When LDL-lowering drug therapy is employed in high-risk or moderately high-risk persons, it is advised that intensity of therapy be sufficient to achieve at least a 30–40% reduction in LDL-C levels.
- For people in lower risk categories, recent clinical trials do not modify the goals and cutpoints of therapy.
Adapted from Grundy et al.8
HMG-CoA reductase inhibitors (statins)
These agents have a powerful LDL-C lowering effect and those currently approved for use differ only in their dose–response curves and unit cost.
Mevastatin was first isolated in 1976 by Endo and colleagues as a natural product from Penicillium species. A related natural product, lovastatin, was approved by the FDA for cholesterol lowering in 1987. Subsequently, simvastatin, pravastatin, fluvastatin, atorvastatin, cerivastatin, and rosuvastatin were developed and approved for use in the US.11 Cerivastatin was withdrawn later because of adverse effects.
Mechanism of action: lipid-altering effects
Brown et al demonstrated that lovastatin inhibits HMG-CoA reductase, the rate-limiting enzyme in cholesterol biosynthesis.13 Statins occupy a portion of the binding site of HMG-CoA reductase, blocking access to the active site of the enzyme.14 Total body cholesterol synthesis is reduced by at least 20%. Ultimately a critical reduction in cholesterol concentration occurs in the liver cell, leading to enhanced production of hepatic LDL receptors15 and increased cellular uptake of LDL-C. Further, reduced very (V) LDL biosynthesis occurs via an effect upon hepatic Apo B secretion, as well as an increased rate of VLDL catabolism.
Pleiotropic effects
In addition to reducing cholesterol biosynthesis, other potential antiatherogenic mechanisms of action for the statins are under current, intense investigation but remain speculative. Pleiotropic effects of statins on the vascular system and the arterial walls – affecting endothelial function, inflammation, coagulation, plaque stabilization, and smooth muscle cell migration – have been identified.16–18 The small GTP-binding protein, Rho, has membrane localization and may mediate the direct vascular effects of statins.
The PRINCE study provides clinical evidence of anti-inflammatory properties of a statin.19 In this prospective, randomized, cohort study, pravastatin lowered levels of C-reactive protein (CRP), an inflammatory biomarker that is predictive of cardiovascular risk. Decreased CRP levels were seen as early as 12 weeks in pravastatin-treated participants (P < 0.001). Pravastatin lowered the median CRP level by 16.9% versus placebo (P < 0.001) at 24 weeks. The decreases occurred in both the primary and secondary prevention groups and regardless of sex, age, smoking, body mass index, baseline lipid levels, diabetes, and use of aspirin or hormone replacement therapy.19
Results of the PPP Project – a meta-analysis of three large, placebo-controlled, randomized trials including almost 20 000 patients and 102 559 person-years of follow-up – provide further clinical evidence that statins may be anti-inflammatory and/or antithrombotic. In particular, statins may be beneficial in reducing strokes.20 Pooled data from two of the trials, CARE and LIPID, involving more than 13 000 patients, showed a 22% reduction in total strokes and a 25% reduction in non-fatal stroke20 The third trial, WOSCOPS, had a smaller trend for reduction in total stroke. Pravastatin reduced the risk of non-hemorrhagic stroke over a wide range of lipid values in patients with documented CHD.20
In the MIRACL trial, atorvastatin (80 mg/day) reduced early recurrent ischemic events in patients with acute coronary syndromes.21 The statin was initiated 24–96 hours after an acute coronary syndrome to over 3000 adults with unstable angina or non-Q-wave myocardial infarction (MI). In the atorvastatin group, 14.8% of patients had a primary endpoint (death, non-fatal acute MI, cardiac arrest with resuscitation or recurrent symptomatic myocardial ischemia requiring emergency rehospitalization) versus 17.4% in the placebo group (P = 0.048).21 The MIRACL investigators suggest that patients with acute coronary syndromes begin statin therapy before hospital discharge, regardless of baseline LDL-C levels.
AVERT compared the efficacy of aggressive cholesterol-lowering therapy versus percutaneous transluminal coronary angioplasty in low-risk, stable patients with CHD. Aggressive lipid lowering was superior to angioplasty in patients with mild to moderate CHD. Atorvastatin was associated with a 36% reduction in ischemic events and a significant delay in time to first ischemic event.22
The CTT study, a meta-analysis of 14 randomized trials using statins in the prevention of CHD, found an approximate 1% reduction in coronary mortality and major vascular events for every 2 mg/dL reduction in LDL-C. These benefits were significant within the first year, but were greater in subsequent years. These results were consistent in all subgroups. There was no evidence that statins increased the incidence of cancer overall, or of any specific site.23
Dosage
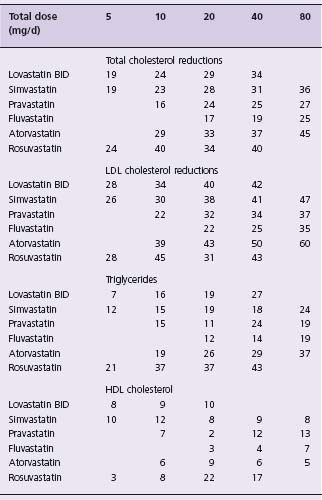
The recommended dosages and described effects of these agents are shown in Table 12.1.11
Impact on lipid levels
All of these agents except atorvastatin and rosuvastatin will lower plasma total cholesterol by 20–40% and LDL-C by 25–45% at maximum approved doses. Triglycerides are reduced by 10–30%. HDL-C plasma levels are frequently increased by 5–10%, but the increases may be more modest or absent in patients with inherently low levels. Lp(a) levels are not affected.11 Statin therapy alters small-dense LDL particles to a larger more buoyant form and also normalizes the responsiveness of coronary vessels to vasoactive stimulus.18
Atorvastatin and rosuvastatin are more powerful members of the statin class. At the maximal dose of 80 mg/day, atorvastatin has resulted in reductions in total cholesterol of 45–50%, LDL-C of up to 60%, triglycerides of 35–45%, and Apo B levels of 35–40%.11 Changes in plasma levels of Lp(a) are small, and increases in HDL-C are inconsistent, but may reach 12%.24 Rosuvastatin is the most recently approved statin and is the most powerful agent of the class on a per mg basis. It has a long half-life (20 hours) and is not metabolized by the cytochrome P450 3A4.25 It has been shown to reduce LDL-C by up to 65%. Atorvastatin and rosuvastatin are somewhat more effective in lowering triglycerides than the other statins, and do so in a dose-dependent fashion.26
Part of the variability of the lipid response, and side effects of the statins, may be due to genetic differences in the rate of drug metabolism. The CYP2D6 phenotype can affect both lipid lowering and tolerability of simvastatin.27 In addition, it has been shown that polymorphisms of the HMG-CoA reductase gene can affect lipid response to statin therapy.28
Adverse reactions
Overall adverse reactions occur in less than 2% of individuals. From 1% to 3% of persons taking a statin will have dose-related, elevated, hepatic enzyme levels.29 While most studies document no abnormalities,1,3,30 most abnormalities that do occur are seen within the first three months of treatment and require monitoring.29 In patients who abuse alcohol, there is an increased risk of hepatic toxicity. An extremely low incidence of adverse events (not significantly different from placebo) has been documented over 5.5 years in the Heart Protection Study (HPS).3
Statins compete with other drugs for specific metabolic pathways of the cytochrome P450 system, whose enzymes act as a major catalyst for drug oxidation in the liver.31 Lovastatin and simvastatin undergo extensive first-pass metabolism by CYP3A4, and caution is urged in using them with cyclosporin (a known inhibitor of CYP3A4), particularly when other inhibitors of the cytochrome P450 system, such as azole-derived antifungal drugs, erythromycin and clarithromycin, are in use, as well as nefazodone and many HIV protease inhibitors. Atorvastatin is also at least partially metabolized by this pathway. Fluvastatin is metabolized mostly by CYP2C9 and few drug–drug interactions have been noted. Pravastatin has less potential for drug interaction with other substrates, inhibitors or inducers of the CYP3A4 and CYP2C9 systems than the other statins because it is metabolized by sulfation, not the cytochrome system.31
About 10% of individuals taking statins may develop myalgias, but perhaps 90% of these symptoms are not related to statin therapy. Statin-induced myalgias and myopathy are most commonly associated with progressive, symmetric symptoms of the hip and shoulder girdles. Many of these patients report muscle pain without concomitant elevations of creatinine kinase (CK). Caution must be used in interpreting CK elevations as they can be increased due to even minor trauma or in sporting activities, and can be caused by other conditions such as hypothyroidism. However, one should consider discontinuing statin therapy if CK increases by more than threefold. Rare (less than 0.1%) and reversible increases of greater than 10-fold in CK levels have been described. The mechanism of muscle injury with statins remains poorly defined, although according to a recent publication it may be related to the HMG-CoA reductase mediated isoprenylation of muscle proteins.32 Statin-associated myopathy, although rare, can progress to rhabdomyolysis.33 This effect can be seen with any statin; however, cerivastatin was voluntarily withdrawn from the world market in 2001 because of an increased rate of rhab-domyolysis compared with other statins.
There are reports of statin use associated with a variety of other potential adverse effects including renal dysfunction, cognitive impairment, increased risk of cancer, peripheral neuropathy, a drug-induced lupus-like state, and cataracts. Despite extensive and widespread use of these agents, no report has established a causal relationship to the use of statins.
Clinical use
Although the biggest proportional reduction in LDL-C levels occurs at low doses, the clinical response to statins is dose dependent, and it appears to be independent of patient characteristics, such as age, gender, smoking status and initial lipid and lipoprotein levels.34 ATP III calls for LDL-C lowering drug therapy in persons with CHD and CHD risk equivalents when the LDL-C is ≥130mg/dL(3.38mmol/L).7 In persons with two or more risk factors for the development of CHD, ATP III suggests that lipid-lowering drug therapy also begin at LDL-C levels ≥130 mg/dL (3.38mmol/L).8 For those at very high risk, a more aggressive LDL-C goal of <70mg/dL (1.82mmol/L) is considered a therapeutic option. This goal should also be considered in those at high risk but with a baseline LDL-C level <100mg/dL (2.60mmol/L).8
ATP III also recommends that LDL-C be measured, either at admission or within 24 hours, in all patients hospitalized with a major coronary event.7 Lipid-lowering drug therapy should be initiated at hospital discharge in a person with a coronary event or procedure if LDL-C is ≥130mg/dL (3.38mmol/L).7 Treatment initiation at hospital discharge takes advantage of patients’ likely higher motivation to comply with therapy at that time and may avoid the “treatment gap” that can occur if outpatient follow-up is less consistent.
In the early 1950s Attschult noted profound reductions in plasma total cholesterol and triglyceride levels in association with use of nicotinic acid. Nicotinic acid has the most marked clinical effect on triglycerides and HDL,35 and is the only lipid-altering agent to consistently lower Lp(a) plasma levels.36 It also can alter small-dense LDL particles to larger, more buoyant forms.36 A complete review of this agent has recently been published.37
Mechanism of action
Nicotinic acid’s predominant effect on plasma lipid levels is to reduce production of VLDL particles, with subsequently reduced production of intermediate-density lipoprotein (IDL) and LDL particles. Nicotinic acid’s major effect on VLDL metabolism results from inhibition of hormone-sensitive, lipase-induced lipolysis in adipose tissue, and decreased triglyceride esterification in the liver. It has recently been demonstrated that niacin exerts its effects by acting as a pharmacologic ligand for the adipocyte and macrophage G protein coupled receptor HM74.39 The physiologic ligand for this receptor is beta-hydroxybutyrate.40 Niacin increases HDL-C to a greater extent than other lipid treatment drugs, and this effect appears to be related to reduced Apo A-I clearance and increased production of Apo A-II.
Dosage
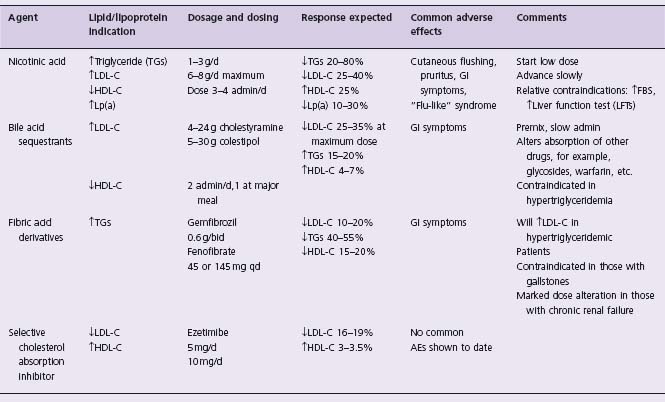
Crystalline nicotinic acid is available in 0.1 and 0.5 g tablets. There is a sustained-release form in dosages of 0.125, 0.25 and 0.5 g and the maximum daily dose is usually 3 g (Table 12.2). A new extended-release form of niacin has relatively mild hepatic effects, and can be taken at bedtime to lessen cutaneous flushing. Extended-release niacin is essentially equivalent to immediate-release niacin in increasing HDL-C. Newer agents that reduce flushing are in development.
Results
Regardless of the patient’s clinical lipoprotein abnormality, dose-dependent reductions in total and LDL-C and plasma triglycerides have been achieved with use of nicotinic acid. HDL-C levels may increase 15–40%; the average increase is 25%, with increases commonly plateauing at a dosage between 1.5 and 3.0 g/d. Reductions in Lp(a) of 25–30% can be achieved.41 As noted above, small-dense LDL particles become larger and more buoyant during nicotinic acid therapy.42
Adverse reactions
Even at very low doses (0.05–0.10 g), nicotinic acid often causes cutaneous flushing (>80%) and pruritus (50%). This effect is due to the subcutaneous release of prostaglandin D2 and is mediated by niacin’s interaction with the G protein coupled receptor HM74 (as is its lipolytic effect, see above).43 There is an ongoing search for agents that block this prostaglandin D2-mediated flushing. In individuals with a history of gout it can precipitate acute gouty attacks. Other frequently noted adverse effects are gastrointestinal symptoms (5–20%), liver enzyme elevations (3–10%, and more common with slow release prerparations), and uric acid increases (5–10%). The clinical picture of mild liver function abnormalities usually resolves with continued therapy or reduced doses.
Some 5–10% of patients taking nicotinic acid will have abnormal glucose tolerance tests or elevated fasting blood sugar levels due to nicotinic acid-induced insulin resistance.44 A flu-like syndrome that can include hepatitis-like findings on liver biopsy, a secretory defect with profound decreases in LDL-C, decreases in HDL-C and a prothrombin time abnormality may occur. This clinical picture is dose dependent and resolves when the agent is stopped.45 Blurred vision with macular edema occurs very rarely.
Clinical use
Many non-prescription forms of nicotinic acid are available in the US and are usually less expensive, but bioavailability may be a problem. Crystalline-form tablets are scored, which allows easy tailoring of the therapeutic regimen, e.g. 0.1 or 0.25 g/d as a starting dose. Dosing with the crystalline form requires three or four administrations a day. No regimen has been shown to be superior to multiples of 0.1 g crystalline tablets administered four times a day. Patients will have little or no effect from two administrations a day, unless using sustained-release preparations. Increases in the dosage are implemented every few days.46 Clinicians commonly reduce the number of administrations to three times per day and use 0.5 g tablets starting with 0.25 g qd for the first week.
Nicotinic acid should always be taken with food, without hot drinks or alcoholic beverages. Dosages should be reduced or perhaps restarted if several successive doses are missed. Cutaneous flushing and pruritus will occur routinely if these precautions are not followed. If symptoms occur, they are the most severe during the first administration. Pretreatment with aspirin or ibuprofen may lessen cutaneous reactions.
Although nicotinic acid may profoundly alter glucose metabolism in some, many diabetic patients have had their lipid disorders successfully managed with this agent. A fasting blood sugar >115 mg/dL predicts subjects who will lose the acute insulin response with an intravenous glucose tolerance test. In patients who have a history of diabetes or glucose intolerance or are at increased risk of diabetes, it is prudent to follow blood sugars closely during initiation of nicotinic acid therapy.
Bile acid-sequestering agents (resins) (see Table 12.2)
This class of agents was first developed for the treatment of cholestasis-related pruritus by Carey and Williams in 1960. Hashim and Van Itallie subsequently demonstrated that cholestyramine lowered plasma cholesterol and it has been in clinical use for 30 years. Other agents in this class are colestipol and the recently approved colesevelam.
Mechanism of action
The enterohepatic circulation of bile acids allows for only 6% or 7% of them to be excreted each day. These polymers have a molecular weight of over 106 and are not absorbed and function by binding bile acids in the gastrointestinal lumen. An increase in bile acid excretion causes an increased production of bile acids in the liver. This results in a relative depletion of cholesterol from the liver cells, thereby inducing inducing an increased level of hepatic LDL receptor activity.47
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


