Introduction and historic perspective
The rapid evolution of the understanding, definition and management of acute myocardial ischemic syndromes constitutes a major milestone of modern cardiology. The widely used terminology of “acute coronary syndromes” (ACS) highlights the specific pathophysiologic mechanisms that distinguish unstable angina (UA), non-ST segment elevation myocardial infarction (NSTEMI), and ST segment elevation myocardial infarction (STEMI) from stable coronary artery disease.1 Pathologic studies have documented the presence of intracoronary thrombus on a ruptured or fissured complex atherosclerotic plaque in 95% of patients with unstable angina who have developed sudden cardiac death.2–4 These plaques are rich in lipids and inflammatory cells and possess a thin cap that makes them prone to rupture.5 At autopsy, the thrombi may be of various ages and at multiple sites; they typically overlie stenotic lesions of only moderate severity; platelet aggregates in the distal small intramyocardial arteries and microscopic foci of necrosis are often found as well.
DeWood documented by angiography that the very early stage of STEMI was consistently associated with a completely occlusive thrombus.6 Subsequent studies focused on the characteristics of culprit lesions associated with ACS, visualized using a variety of methods including angiography, intravascular angioscopy, ultrasound, and thermography. It became recognized that the active plaque was often not unique, suggesting a more diffuse inflammatory state. These concepts led to a new era of research in cell biology and clinical investigation supported by emerging technologies and methodologically sophisticated clinical research, providing a foundation for our current evidence-based approaches to therapy.
New dimensions and definitions
The definitions of ACS encompass various aspects of the disease including epidemiology, prognosis, clinical manifestations, pathophysiology, and therapeutic options. Their delineation has led to improved understanding of the mechanisms and manifestation of plaque activation, and has opened new therapeutic perspectives. Professional practice guidelines for the management of UA and NSTEMI were first published in 1979, jointly by the American College of Cardiology and the American Heart Association and independently by the European Society of Cardiology. They were subsequently updated in 2002 and 2007 to incorporate algorithms for rapid diagnosis, risk stratification, patient triage, and therapy.1,7 Large registries8–10 monitor national and international adherence to guidelines and their performance in various hospital settings as well as their impact on quality of care and prognosis. CRUSADE has been succeeded by the American College of Cardiology’s NCDR-ACTION Registry™ (Acute Coronary Treatment and Intervention Outcomes Network), which collects data from hundreds of hospitals across the US into one unified platform with standardized clinical data elements to facilitate benchmarking outcomes, and to analyze treatment regimens.11 Such initiatives are leading the way in the monitoring and evaluation of quality of care and are facilitating the systematic application of guidelines.
From a clinical perspective, a fundamental aspect of an acute coronary syndrome is the recognition of a change in the pattern of ischemic symptoms to one of increased severity. In the absence of evidence of causative extracoronary factors, such symptoms may signal a rapid progression in the severity of the coronary artery disease. As this situation may demand immediate attention, any healthcare professional should be able to make a diagnosis of possible ACS on first seeing the patient. The term “acute coronary syndrome” has thus become a working diagnosis which facilitates rapid access to diagnostic and therapeutic procedures. It provides a logical framework within which to categorize and risk stratify patients who present with a constellation of clinical symptoms compatible with acute myocardial ischemia. It also encourages consistent diagnostic terminology and patient selection for clinical trials and epidemiologic studies. The working diagnosis of ACS is based upon clinical and ECG characteristics which guide immediate management. The subsequent clinical course and appropriate laboratory tests will lead to a final diagnosis which could be ST segment elevation myocardial infarction (STEMI), non-ST segment elevation myocardial infarction (NSTEMI), unstable angina, stable angina or another cardiac or non-cardiac condition (Fig. 29.1).
Figure 29.1 Nomenclature of acute coronary syndromes (ACS). The spectrum of clinical conditions that range from unstable angina to non-Q wave MI and Q-wave MI is referred to as acute coronary syndrome. Patients with ischemic discomfort may present with or without ST segment elevation on the ECG. Most patients who present with non-ST segment elevation ACS will eventually be classified as having unstable angina or non-Q wave MI. The distinction between these two diagnoses is ultimately made based on the presence or absence of a cardiac marker detected in the blood. Only a minority of patients with non-ST elevation ACS will develop a Q-wave MI. Most patients with ST segment elevation ACS will evolve to develop a Q-wave MI, although some may evolve to a non-Q wave MI or may occasionally resolve without myocardial necrosis. (Adapted with permission from Anderson JL, Adams CD, Antman EM et al.1)
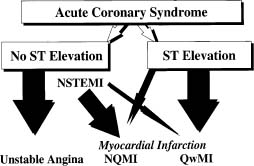
Table 29.1 summarizes the clinical manifestations and Figure 29.1 provides current nomendature for ACS. An immediate 12-lead ECG must be obtained, and repeated when the pattern of pain changes, to detect ST elevation or new left bundle branch block (LBBB) indicative of evolving STEMI and the need for immediate reperfusion therapy.13 In the absence of ST segment elevation, the working diagnosis becomes a non-ST segment elevation acute coronary syndrome, the likelihood of which is increased by the presence of ST segment depression and/ or T-wave inversion on the ECG.1 An elevated cardiac troponin T or I level then indicates an evolving NSTEMI, while normal values point to unstable angina. If troponin assays are not available, the best alternative is CK-MB measured by mass assay.
Table 29.1 Clinical presentation of non-ST segment elevation acute coronary syndrome
New-onset angina | New-onset angina of at least CCS class III in severity12 I. Angina at strenuous, rapid or prolonged exertion. II. Slight limitation of ordinary activity, e.g. angina occurs on walking or climbing stairs rapidly. III. Marked limitation of of ordinary activity; angina occurs on walking 1-2 blocks on level or climbing 1 flight of stairs in normal conditions at normal pace. IV. Inability to perform any physical activity without discomfort; anginal syndrome may be present at rest. |
Increasing (crescendo) angina | Angina that has become distinctly more severe, more frequent, longer in duration, or lower in threshold (that is, increased by greater than or equal to 1 CCS class to at least CCS class III severity) |
Rest angina | Angina occurring at rest and prolonged, usually > 20 minutes |
Early post-MI ischemia | Ischemic chest pain recurrent within 30 days after MI |
The availability of troponins T and I has increased the sensitivity of the diagnosis of myocardial infarction and has sharpened the distinction between unstable angina and myocardial infarction.1 Troponin measurements allow the detection of cell necrosis and myocardial infarction in up to 30% of patients who would otherwise be diagnosed as having unstable angina based on normal CK-MB blood values, ultimately improving patient care and outcomes.13 On the other hand, although highly sensitive and specific for myocardial cell necrosis, cardiac troponins are not specific for cell death related to ischemia resulting from coronary artery disease. The latest expert consensus document for a universal definition of myocardial infarction14 lists the potential causes of troponin elevation outside the context of ischemia; some of them are commonly found in the age range of patients with an ACS, e.g. congestive heart failure, aortic valve disease, tachy or brady arrhythmias, renal failure, and pulmonary embolism. Myocardial infarction is defined as myocardial cell death due to prolonged myocardial ischemia. Myocardial infarction may be diagnosed when a rise and/or fall of cardiac biomarkers (preferably troponin) is detected, with at least one value above the 99th percentile of the upper reference limit, together with evidence of myocardial ischaemia with at least one of: i) symptoms of ischaemia; ii) ECG changes indicative of new ischemia (new ST-T changes or new LBBB); iii) development of pathologic Q-waves in the ECG; or iv) imaging evidence of new loss of viable myocardium or new regional wall motion abnormality. Detection of a rising and/or falling pattern of troponin is needed to distinguish MI from elevated background levels; but this pattern is not absolutely required for the diagnosis of MI in a patient presenting > 24 hours after the onset of symptoms because troponin values may remain elevated for 7–14 days following the onset of symptoms. Thus, troponin elevation is an indicator of MI, usually STEMI when there is ST segment elevation or a left bundle branch block and NSTEMI when there is not. True posterior or high lateral MIs can be electrocardio-graphically silent. Furthermore, the absence of ST segment shifts on the admission ECG does not rule out an acute coronary syndrome. Serial ECGs are required and cardiac markers must be measured 6–9 hours after the onset of pain when the first determination is negative. A negative work-up may not totally exclude ACS or even coronary artery disease and the need for treatment, but generally does confer a favorable prognosis.
A quick appraisal of the patient’s demographics, risk factors, cardiac and non-cardiac antecedents, and medications is mandatory for decision making. The characteristics of chest pain and associated symptoms complement the evaluation of disease severity. Thus new-onset angina, crescendo or progressive angina, pain at rest, nocturnal pain, prolonged pain, and recurrent ischemia after myocardial infarction carry increasingly worse prognoses. Although the Canadian Cardiovascular Classification12 was designed to describe the severity of stable exertional angina, in practice it is often applied to describe angina of sufficient progression and severity to warrant the designation of ACS.
Incidence, natural history and prognosis
There were 4 497 000 visits to US emergency departments (EDs) with a primary diagnosis of cardiovascular disease (CVD) in 2003.15 The National Center for Health Statistics reported 1 565 000 hospitalizations for primary or secondary diagnosis of an ACS in 2004, 669 000 with UA and 896000 with MI.15 Statistics on NSTE-ACS are less reliable than those of STEMI because the diagnostic criteria have been evolving over time, with the introduction of more sensitive and specific criteria allowing the identification of more patients with the diagnosis but limiting the hospitalization to higher-risk patients. On the other hand, the absolute incidence of STEMI has been decreasing for various reasons that are likely related to better control of risk factors and use of drugs effective in primary prevention.16 Reperfusion therapy and antithrombotic therapy have also dramatically reduced the short-and long-term mortality and morbidity associated with STEMI. Such is not the case in NSTEMI, which is often associated with more risk factors, including older age, and with a more advanced and diffuse atherosclerotic process. Accordingly, NSTEMI is now associated with a worse prognosis than STEMI past the acute phase. The average age of a person having a first heart attack is now 65.8 years for men and 70.4 years for women; 43% of ACS patients of all ages are women.15
Epidemiologic data using the WHO MONICA criteria for the diagnosis of Q-wave MI showed a more than 30% decrease in the mortality rates from Q-wave MI between 1975 and 1995, two-thirds of this decline being attributable to reduced incidence and one-third to decreased hospital mortality.17–20 The ENACT registry, which included 3092 patients from 29 European countries in the mid-1990s, showed that the distribution of admission diagnosis was unstable angina/NSTEMI in 46%, myocardial infarction in 39%, and suspected ACS in 14%, with no regional differences across Europe.21 In the more recent Global Registry of Acute Coronary Events (GRACE) that collected data from 10 693 patients across North and South America, Australia, New Zealand, and Europe between 1999 and 2001, two-thirds of admitted patients had unstable angina/ non-ST segment elevation ACS and one-third STEMI.22 This ratio is reversed in India, with its low socio-economic status.23
In an epidemiologic study of 5832 residents from metropolitan Worcester, Massachusetts, the annual incidence of Q-wave MI between 1975 and 1997 decreased from 171 per 100 000 population to 101 per 100 000 compared with an increase from 62 to 131 per 100 000 for non-Q wave MI.24 The hospital mortality for Q-wave MI in the registry declined from 24% to 14%, but that of non-Q wave MI remained constant at 12%. Similarly in the GRACE registry, the risk-adjusted hospital deaths declined between 1999 and 2006 by 18% in STEMI and 0.7% in NSTEMI.25 It appears that despite impressive declines in the incidence and in-hospital and long-term mortality rates of Q-wave MI, the incidence of non-Q wave MI has been stable or increasing with mortality rates that have changed little over the last decade. Part of these data in NSTEMI might be explained by the wide use of troponin resulting in higher sensitivity and specificity for the diagnosis, and selective hospitalization of higher risk patients.
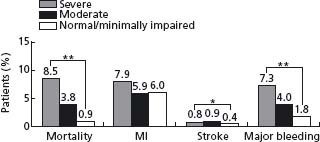
The natural history of UA/NSTEMI is difficult to assess given the increasingly effective therapeutic interventions introduced over the last 2–3 decades in parallel with better diagnostic and risk stratification tools. Treatment now targets multiple facets of the disease, combining drugs and interventions. Early studies have shown a 10-fold increase in the short-term risk of an event in patients with unstable angina compared with patients with stable angina, and several-fold more compared with individuals with risk factors but no known coronary disease. Recent randomized trials have shown an impressive decrease in MI and death. Prior to the routine prescription of bed rest, nitrates and beta-blockers for unstable angina, the one-month rate of MI was in the range of 40%, and of death, 25%.26 By the 1970s these rates had fallen to about 10% and 2%. In 1979–80, a study of all patients hospitalized with unstable angina in Hamilton, Canada, over a period of one year noted showed in-hospital and one-year mortalities rates of 1.5% and 9.2% respectively.27 By the time of the studies with heparin in the late 1980s, study inclusion criteria had shifted toward patients at somewhat higher risk with some trials limiting enrollment to patients with NSTEMI. The rates of the composite outcome of death or non-fatal myocardial infarction by five days were then about 10%.28,29 They fell to about 4% with the addition of heparin to aspirin, and further with enoxaparin and the glycoprotein IIb/IIIa antagonists. The event rates at 30 days in recent trials with new antithrom-botic therapies and an invasive management strategy are shown in Figure 29.2.30–41 The risk of the disease is highest in the first few days, decreases over the following weeks and months, and eventually becomes similar to the prognosis of patients with stable angina. The long-term prognosis is influenced by the severity of the underlying disease. In the OASIS registry, the incidence of events was 10% at one month and increased steadily in the following two years to reach more than 20% after 24 months (Fig. 29.3), and was still higher in diabetic patients and in patients with previously known coronary artery disease.42 In the GUSTO-II study, the in-hospital mortality rate was highest, as expected, in patients with STEMI; however, increasing mortality during follow-up in patients with ST segment depression exceeded that of patients with ST elevation after six months, reaching 8.7% vs 6.8% (Fig. 29.4).43
Figure 29.2 Rates of death and of death or myocardial infarction in contemporary trials that evaluated new antithrombotic drugs and routine invasive treatment strategy in patients with non-ST segment elevation ACS. Data are the average of events in the intervention and control groups. The interventions resulted in a reduction in rates of death ranging from–5% to 36% and in rates of death or myocardial infarction ranging from–8% to 27%.29–40
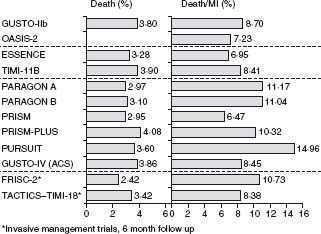
Figure 29.3 Kaplan–Meier event curves for diabetic and non-diabetic patients for total cardiovascular death, MI, stroke, and new onset of congestive heart failure during a 24-month follow-up period after an episode of non-ST segment elevation ACS. (Data from the OASIS Registry study reproduced with permission from Malmberg et al.42)
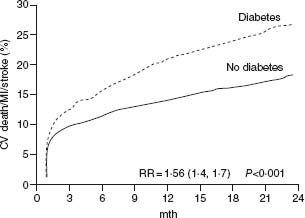
Figure 29.4 Kaplan–Meier estimates of probability of death at six months by ECG changes at admission. The event rate was highest in-hospital in patients with ST segment elevation MI. However, by six months, mortality in patients with ST depression exceeded that of patients with ST elevation. (Data from the GUSTO-II study, reproduced with permission from Savonitto et al.43)
The empiric concept that NSTEMI represents an unresolved ACS at risk of completion, likely due to an underlying disease process that remains active, appears to be correct for a significant number of patients. This is supported by data showing that markers of inflammation and of activation of coagulation may remain elevated months past the acute phase of an episode of ACS.44,45
A gradient in risk exists in ACS from relatively benign to severe; accordingly, patient management should be guided by clinical risk stratification. The high-risk features for death and ischemic events present at admission, or developing rapidly, have been identified in many registries and clinical trials. Registry studies look at a broad spectrum of patients with acute chest pain and are likely to provide more “real-world” data22,24,42 while clinical trials enroll more selected and homogeneous populations predefined by entry criteria, collect data prospectively, and often include innovative substudies to test new hypotheses. The two approaches are complementary.
Cardiac risk factors are strong predictors of the presence and prognostic of coronary artery disease, but are generally not useful for the diagnosis of an ACS and its associated risk.46 Angiographic predictors of a future episode of ACS are a known high extent score of the disease defined by the number of coronary artery segments showing a stenosis whatever its severity.47 On the other hand, the occurrence of an ACS is a strong indicator that the severity of an obstructive lesion has progressed significantly.48 Many parameters influence prognosis, some related to the acute thrombotic events, others to the extent and severity of the underlying disease, and still others to co-morbidities.
The clinical evaluation, the 12-lead ECG, and the tropo-nin T or I blood levels are powerful and now standard first-line instruments for risk evaluation. These are included in risk scores, are part of recommended treatment algorithms, and are used as entry criteria in clinical trials to identify the high-risk patients. There are, however, other strong demographic and clinical predictors of prognosis that impact on the prognosis of UA and that should now also be considered in risk stratification. These are mainly older age, left ventricular function, and co-morbid conditions, particularly renal failure and diabetes. Bleeding has recently emerged as a strong prognostic factor; it is largely iatrogenic as a result of aggressive treatment.
In the evaluation of prognosis, the physician is first oriented by symptoms, recognizing an increasingly severe prognosis from new angina, to progressive angina, rest angina, and prolonged chest pain, further worsened if ischemic episodes are accompanied by hemodynamic or electrical instability and if they are recurrent despite optimal medical therapy.1 It is important to recognize that women, the elderly, and the diabetic patient are more likely to have atypical presentations, because the prognosis with atypical symptoms at the time of an infarction can be worse than that of patients with more typical symptoms.49 Women enrolled in clinical trials are in general older than men and have more risk factors such as diabetes and hypertension. The proportion of women with ST segment elevation is less than in men but their prognosis is then worse. Women who present with NSTE-ACS less often have an elevation of the cardiac markers and have a better prognosis. The odds ratio (OR) for infarction and death in the GUSTO-IIb study in women compared to men was 0.65 (95% confidence interval (CI) 0.49 –0.87; P = 0.003),50 and in the non-invasive strategy arm of the FRISC-II study was 0.64 (95% CI 0.43–0.97; P = 0.03).51 Coronary angiography in general revealed less severe coronary artery disease among women than men.51
Diabetes
Diabetes has become a major determinant of risk as its prevalence has reached epidemic proportions. It is present in 20–25% of patients enrolled in trials in ACS. Diabetes increases morbidity and mortality in the setting of ACS and after percutaneous interventions and coronary artery bypass grafting. In the OASIS registry, diabetes was an important and independent predictor of two-year mortality (relative risk (RR) 1.57; 95% CI 1.38–1.81; P < 0.001), as well as of cardiovascular death, new myocardial infarction, stroke, and new congestive heart failure.42 The relative risk of death in diabetic women was significantly higher than in diabetic men (RR 1.98 and 1.28 respectively). Diabetic patients without known prior cardiovascular disease had the same event rates for all outcomes as non-diabetic patients with previous vascular disease (see Fig. 29.3). In a subgroup analysis of patients enrolled in 11 independent TIMI Group clinical trials from 1997 to 2006 that included 62 036 patients (46 577 with STEMI and 15 459 UA/ NSTEMI), 17.1% had diabetes.52 The unadjusted 30-day mortality rates were significantly higher among patients with diabetes than those without for both UA/NSTEMI (2.1% vs 1.1%, P < 0.001) and STEMI (8.5% vs 5.4%, P < 0.001), as were the odds ratios adjusted for baseline characteristics and management (1.78; 95% CI 1.24–2.56 and 1.40, 95% CI 1.24–1.57 respectively). The hazard ratio (HR) for one-year mortality after UA/NSTEMI was 1.65; 95% CI 1.30–2.10) and after STEMI (HR 1.22; 95% CI 1.08–1.38). By one year following ACS, patients with diabetes presenting with UA/NSTEMI had a risk of death that approached that of patients without diabetes presenting with STEMI (7.2% vs 8.1%).
Depression may also have a negative impact on prognosis. In a study of 430 patients with NSTE-ACS, depression predicted the endpoint of cardiac death or non-fatal MI, with an adjusted odds ratio of 6.73 (95% CI 2.43 –18.64; P < 0.001) after controlling for other significant prognostic factors that included baseline ECG, left ventricular ejection fraction, and number of diseased coronary vessels.53
Chronic kidney disease
Chronic kidney disease (CKD) is associated with a higher risk of cardiovascular and all-cause mortality.54–56 It is also a marker of impaired prognosis in patients with coronary artery disease or with a myocardial infarction, and in patients with an ACS (Fig. 29.5) or undergoing coronary angiography. These cross-interactions are likely linked to a high incidence of traditional risk factors in CKD, mainly diabetes and high blood pressure, and also of non-traditional risk factors such a proinflammatory state, a pro-thrombotic state, and hyperhomocysteinemia. Complicating these issues, CKD is associated with a higher bleeding risk (see Fig. 29.5); the more severe the dysfunction, the higher is the bleeding risk.57 Routine calculation of the estimated creatinine clearance rate by the Cockcroft–Gault method or of the estimated glomerular filtration rate (GRF) by the Modification of Diet in Renal Disease (MDRD) formula is strongly recommended over the simple measure of plasma creatinine levels.58 Cystatin C is also emerging as a valid marker of GFR.59
Figure 29.5 Hospital outcomes according to degree of renal impairment for the subgroup of patients with non-ST-elevation myocardial infarction/unstable angina from the GRACE Registry. * P < 0.05 across all categories of renal function; * * P < 0.0001 across all categories of renal function. (Reproduced with permission from Santopinto et al.59.)
The 12-l ead ECG
Current information on the prevalence of ECG abnormalities is difficult to obtain, in part because the ECG and other diagnostic criteria used to select patients for clinical registries are often less homogeneous and rigorous than those used for clinical studies. In the TIMI-III Registry of 1416 patients enrolled because of unstable angina or non-Q wave MI,60 ST segment deviation >1 mm was present in 14.3% of patients, isolated T-wave inversion in 21.9%, and LBBB in 9.0%. By one-year follow-up, death or MI had occurred in 11% of patients with ST segment depression, 6.8% of patients with isolated T-wave inversion, and in 8.2% of those with no ECG changes. ST segment depression 0.5 mm or more and LBBB were significant predictors of death and MI, with rates of 16.3% and 22.9%, respectively. The ECG is not infrequently confounded by LBBB, left ventricular hypertrophy, paced rhythm or other derangements. In the PARAGON-A study, these confounders were associated with near doubling in the one-year mortality rates (12.6% versus 6.5%). Among the 12 142 patients enrolled in the GUSTO-II trial with symptoms at rest within 12 hours of admission and ischemic ECG changes, 22% had T-wave inversion, 28% ST segment elevation, 35% ST segment depression, and 15% ST segment elevation and depression.43 The 30-day rates of death or myocardial reinfarction were 5.5%, 9.4%, 10.5%, and 12.4% respectively (P < 0.001). The cumulative rates of death in this study are shown in Figure 29.4.
There is a gradient of increasing risk of death or myocardial infarction in hospital and up to one year, from non specific ECGs to T-wave inversion to ST segment depression including confounding ST-T changes. Such a gradient exists from ST segment depression >0.05mm, to >1 mm, to >2mm, to >2 mm, and to depression in more than two contiguous leads.61 The prognostic value of ST segment depression extends to four years following hospital discharge.62 Special attention is required for patients showing deep T-wave inversions in leads V1 through V6 and in leads I and AVL on the admission or on subsequent ECGs even in the absence of symptoms; the changes are quite specific for the presence of significant disease in the proximal left anterior coronary artery and are predictive of a high risk of progression to an infarction that can be massive; when the T-wave inversions spare leads I and AVL, the mid-LAD is more often the site of the culprit lesion.63 Similarly, ST segment elevation in lead AVR associated with ST-T changes in other leads can be a marker of left main disease and severe three-vessel disease. The two last criteria deserve special attention.
The importance of recording the 12-lead ECG during chest pain must be emphasized. The detection of ST segment depression during pain has diagnostic and prognostic value.64 Occasionally, transient ST segment elevation will be detected in association with a critical dynamic coronary artery stenosis due to spasm or thrombus formation, or with small vessel disease. ST segment shifts during pain occurring on medical management indicate refractory ischemia, an endpoint commonly used in clinical trials. Such refractory ischemia predicts near tripling of adjusted one-year mortality.65
Biomarkers
Markers of necrosis
Elevation of serum troponin follows the ischemic insult by six hours, as does CK-MB; with newer assays, troponin elevation may be detected earlier. Myoglobin can be useful as an early and sensitive marker of necrosis as it rises within two hours after the onset of pain to peak within 4–6 hours; however, it is non-specific and mandates confirma-tion of the cardiac origin by elevation of CK-MB or troponin. CK-MB is a valid second choice to troponin when determined by a mass assay. The measurement of ancillary markers such as total CK, AST, ALT is unnecessary and can be misleading.
In contrast to CK-MB and myoglobin, cardiac troponins T and I are not normally detectable in the peripheral blood and therefore provide an exquisitely sensitive and specific marker of cardiomyocyte necrosis. The necrosis is usually of ischemic origin, but there are multiple causes of myocardial necrosis other than MI. Only one elevated value is required for the diagnosis of myocardial infarction as long as the patient presents within 24 h after the onset of compatible symptoms. Otherwise, such as in patients with renal failure, the diagnosis of acute MI requires the demonstration of a rising and/or falling pattern against the background value.
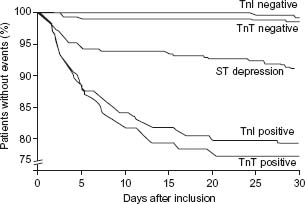
Multiple studies have validated the prognostic value of an elevation in the serum troponin levels.66 Figure 29.6 depicts the results of a clinical trial of a study of patients consulting in the emergency department for acute chest pain;18 the 30-day rate of death or myocardial infarction was highest in patients with elevated troponin T or troponin I levels, intermediate in patients with ST segment depression, and lowest in patients with normal troponin levels. The higher the elevation in troponin levels,67 the worse the prognosis, but even small elevations are associated with a significantly impaired prognosis.68 In the FRISC study, among patients with a non-ST segment elevation ACS, the risk of myocardial infarction or cardiac death at six months was respectively 4.3%, 10.5%, and 16.1% in patients within the first, second, and third tertile of maximal elevation of troponin during the first 24 hours.69,70
Figure 29.6 Risk of death or non-fatal myocardial infarction during 30 days of follow-up by troponin T (TnT) and I (TnI) levels, elevation (positive) or no elevation (negative), and ST segment depression on the ECG. The risk for myocardial infarction and death increases with increasing serum troponin concentrations and may be 20% in 30 days and 25% within six months in patients with the highest troponin levels. (Reproduced with permission from Hamm and Braunwald.70)
A meta-analysis among patients with unstable angina, including 12 reports with troponin T and nine with troponin I, demonstrated a risk ratio for the occurrence of myo-cardial infarction at 30 days of 4.2 (95% CI 2.7–6.4; P < 0.001) for troponin I and of 2.7 (95% CI 2.1–3.4; P < 0.001) for troponin T.71 A second meta-analysis included 18982 patients with unstable angina from 21 studies and showed odds of death or myocardial infarction at 30 days of 3.44 (95% CI 2.94–4.03; P < 0.00001) for the total population of troponin-positive patients, 2.86 (95% CI 2.35–3.47; P < 0.0001) for patients with ST segment elevation, 4.93 (95% CI 3.77 –6.45; P < 0.0001) for patients with non-ST segment elevation, and 9.39 (95% CI 6.46 –13.67; P < 0.0001) for patients with unstable angina.72 A third meta-analysis included seven clinical trials and 19 cohort studies. The odds of mortality among 11 963 patients with positive tro-ponin T or I was 3.1 (5.2% versus 1.6%). The discriminative value of elevated troponin levels was greater in cohort studies than in clinical trials: 8.4% versus 0.7% (OR 8.5) for troponin I, and 11.6% versus 1.7% (OR 5.1) for troponin T.73
Determination of troponin levels has many utilities. Elevated levels in the setting of NSTE-ACS strongly suggest the presence of an active intracoronary thrombotic process that is associated with small foci of myocardial necrosis, likely because of distal embolization of thrombotic material originating from the culprit lesion. Beyond providing a highly sensitive and specific test for the diagnosis of myo-cardial infarction, any elevation provides important prognostic information in acute coronary syndromes. Patients with troponin elevation are also more likely to profit from therapy with a Gp IIb/IIIa antagonist,74 from a low molecular weight heparin,75 and from interventional procedures.76 Some have recently questioned the prognostic significance of the very small elevation of troponin that can be detected with the new generations of ultrasensitive tests.
Markers of inflammation
Consistent with excessive plaque inflammation as the main trigger to plaque rupture and thrombus formation, a wide range of markers of inflammation are found to be elevated in ACS. These include acute phase proteins (C-reactive protein, serum amyloid A protein, fibrinogen), soluble adhesion molecules (sVCAM-1, sICAM-1, E-selectin, P-selectin), proinflammatory cytokines (interleukin-6, TNF-alpha, interleukin-18), degradation enzymes matrix metalloproteinases, placenta-associated plasma protein-A), and markers of thrombosis (D-dimers, F1.2), oxidative stress (myeloperoxidase), inflammation (CD40 ligand), and of immune system activation (neopterin).
C-reactive protein (CRP) is a non-specific but highly sensitive marker of an inflammatory state. It is produced by the liver upon stimulation by interleukin-6, which is induced by TNF-alpha, IL-1, IL-18, platelet-derived growth factor, antigens, and endotoxins. As CRP half-life is 19 hours, blood levels are determined mainly by rates of production, These can be very high in inflammatory and infectious disease, and in the acute phase of a large inf(myeloperoxidase,arction where they rise within six hours and peak at 48 hours. In NSTEMI, the elevation precedes that of markers of myocardial necrosis in patients who had previous progressive angina, but not in patients without.77 The early levels may thus be less useful for risk evaluation; on the other hand, levels obtained after 24–48 hours can represent a transient non-specific acute phase reaction caused by the ongoing cell necrosis. and inconsistently predict future events. Elevated levels at a later time and before hospital discharge are found in up to 40–50% of patients and are associated with high rates of late cardiac events, including death/MI/ recurrent ischemia at 12 months,78 death/MI at six months79,80 and up to two years,81 and death at 36 months.82 In the CAPTURE trial of 447 patients, CRP levels >10mg/L did not predict mortality or myocardial infarction at 72 hours in contrast to elevated troponin T levels, but did predict death or MI at six months (18.9% compared to 9.5%), independently of the troponin status.80 Of interest, high levels of neopterin levels have been linked with the presence of multiple active inflammatory plaques.83
The assessment of CRP levels is not currently part of the recommendations of various guidelines. The cut-off points that best predict early and late prognosis as well as the ideal timing for blood sampling remain to be better defined and no study has yet documented that a treatment specific for CRP has improved prognosis. In one study, PCI or CABG had little effect on the one-year excess of recurrent ischemic events in patients with NSTE-ACS and high CRP levels. Elevated CRP levels are associated with increased risk of restenosis and acute complications after PCI,84,85 and with an increased risk of new ischemic events up to eight years after CABG.86 The JUPITER trial was designed to determine whether long-term treatment with rosuvastatin (20 mg orally od) would reduce the rate of first major cardiovascular events, defined as the combined endpoint of cardiovascular death, stroke, myocardial infarction, hospitalization for unstable angina, or arterial revascularization among individuals with LDL-C levels <130mg/dL (3.36 mmol/L) at high vascular risk because of an enhanced inflammatory response as indicated by hsCRP levels ≥2mg/L.87 The trial was terminated prematurely upon the recommendation of the DSMB because early efficacy.
Beta-type natriuretic peptides
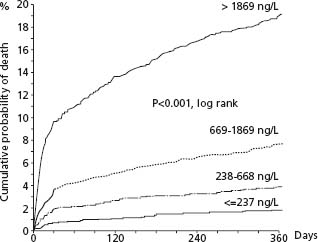
Beta-type natriuretic peptides are cardiac neurohormones released in response to ventricular myocyte stretch; proBNP is enzymatically cleaved to the N-terminal proBNP (NTproBNP) and subsequently to BNP. The value of assessing BNP was first shown for the diagnosis and evaluation of heart failure. Since then, large data sets have documented a high predictive value independent of conventional risk factors and other biomarkers in patients with STEMI and with UA/NSTEMI.88,89 This predictive value extends to early and late blood sampling. Increasing levels are associated with proportionally higher short-and longterm mortality rates. In the GUSTO-IV trial of 6809 patients, the one-year mortality rates by increasing quartiles were 1.8%, 3.9%, 7.7%, and 19.2%, respectively (P < 0.001). (Fig. 29.7) This prognostic value was independent of a previous history of HF and of clinical or laboratory signs of LV dysfunction on admission or during hospital stay. A previous study had shown that this prognostic value was applicable to patients with unstable angina, STEMI, and particularly NSTEMI. The exact reasons for this selective predictive value in ACS are not clear but likely relate to larger zones of ischemic myocardium associated with necrosis and stunning and is in line with the impaired prognosis shown with modest depression of LVEF.90 Current guidelines do not recommend routine determination of natriuretic factors because elevations would be unlikely to prompt the use of specific evidence-based therapies, and the prognostic value lies more in long-term than in short-term outcomes. It is stated as a class II recommendation, however, that natri-uretic factors may be measured to supplement the assessment of global risk in patients with suspected ACS.1 They should also be used if another indication exists.
Figure 29.7 Patients with acute coronary syndromes without ST segment elevation in GUSTO-IV. Kaplan–Meier survival curves regarding probability of death during 1 year for patient strata, according to quartiles of NT-proBNP. (Reproduced with permission from James et al89.)
Multiple markers and panels of markers
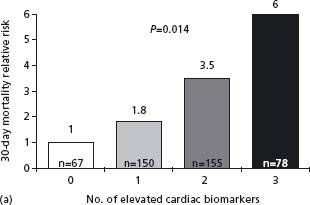
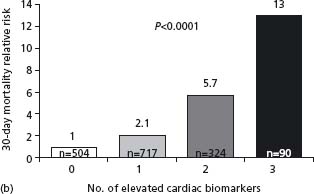
The mechanisms of ACS are highly complex and involve many pathophysiologic cascades with numerous interactions between them. Many studies have shown that a multimarker approach will improve the prognostic value. In the TIMI-11A study of 630 patients with a non-ST segment elevation ACS, the risk of death at 14 days was highest with elevated troponin T and CRP, intermediate when either marker was elevated, and lowest when both were normal (CRP < 1.55mg/L).91 In the GUSTO study, levels of NT-proBNP, troponin (Tn) T, CRP, heart rate, and creatinine clearance, in addition to ST segment depression, correlated independently with one-year mortality, but NT-proBNP was the marker with the strongest relation.89 In contrast, only troponin T, creatinine clearance, and ST segment depression were independently related to future MI. The combination of NT-proBNP and creatinine clearance provided the best prediction, with a one-year mortality of 25.7% with both markers in the top quartile vs 0.3% with both markers in the bottom quartile. The cumulative benefit of using TnI, CRP and BNP was demonstrated in substudies of two large clinical trials92 (Fig. 29.8). Multimarker measurement is not recommended at the present time since the cost/benefit ratio would be low in the absence of a prospectively validated scenario. Troponin assessment is a must–another marker and the only one recommended for routine use. BNP or NT-proBNP could be useful in some patients for a more global evaluation and CRP as well.
Figure 29.8 Relative 30-day mortality risks in OPUS-TIMI 16 (a) and TACTICS-TIMI 18 (b) in patients stratified by the number of elevated cardiac biomarkers. (Reproduced with permission from Sabatine et al92.)
The near future will see the introduction of growing numbers of markers to help delineate the complex and dynamic pathophysiology of cardiovascular diseases and tailor treatment selection to specific patient needs. These will include clusters of blood markers tracking various pathophysiologic pathways, cell membrane markers, the genome and proteome, and molecular probes for tissue imaging.
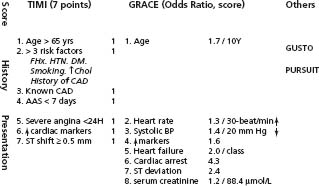
Prognosis can be predicted by various clinical, ECG, and laboratory parameters. Accordingly, predictive models have been derived from various databases by applying multiple regression analyses to identify independent predictors of prognosis. However, the results of such analyses are influenced by the characteristics of the test populations and by the baseline data collected. The GUSTO, TIMI and PURSUIT scores93 were developed from the databases of large clinical trials of NSTE-ACS, whereas the GRACE score was built on registry data acquired across the entire spectrum of ACS. The TIMI score excluded patients with planned revascularization within 24 h and those at high bleeding risk from enoxaparin treatment (including significant renal dysfunction); it looked at the rates of death/MI and recurrent ischemia requiring an intervention by 14 days. Renal failure was an exclusion criterion in the PURSUIT trial, and the outcome of death or MI was assessed at 30 days. The GRACE score was shown to be useful to predict death or MI at one year and death at six months and four years.94,95 It can be measured at admission, to assess the risk of death or death or MI from admission to discharge or to six months, or at hospital discharge to evaluate the risk to six months or to one year. These scores all add to the prognostic value of biomarkers and the ECG and are useful to identify patients who will most benefit from more aggressive medical and invasive treatment.96
Age is a strong predictor in all scores. The TIMI score can be readily and simply assessed at admission or shortly thereafter by calculating the sum of seven independent predictors, allowing discrimination of a 10-fold difference in risk through 14 days (Fig. 29.9). The PURSUIT and GRACE scores added an important dimension of LV function and the GRACE score a component of chronic renal disease.
Figure 29.9 Risk scores used in UA/NSTEMI. The components of the TIMI and GRACE scores are shown in detail. Each component in the TIMI score counts for 1. Odds ratios are given for the components of the GRACE score. Algorithms to calculate these scores can be easily downloaded from the Web for use in bedside devices.
This section will focus on the mechanisms of acute coronary syndromes that are the most relevant with respect to management. Interested readers are referred to more exhaustive reviews.5,97 The culprit lesion becomes clinically manifest only with the development of an obstruction severe enough to impede coronary blood flow at rest, or when it is the site of a thrombotic occlusion shedding thromboembolic material into the distal coronary circulation. Therefore, the active plaque is clinically detected only at an advanced stage of the underlying disease. Further, the concept of a single active plaque has been challenged. Pathologic studies have shown multiple rupture sites and thrombi at multiple sites often associated with platelet aggregates in small intramyocardial arteries and microscopic foci of necrosis.4,98,99 An angiographic study in 253 patients with an acute myocardial infarction documented that complex and ruptured plaques could be found in 40% of patients and that these were associated with a 10-fold increase in the risk of a recurrent ACS.100
Atherosclerosis is the substrate for ACS. The severity of atherosclerosis underlying an episode of an acute coronary syndrome is highly variable, ranging from absence of significant stenoses to the presence of left main disease in 5–10% of patients, and single-, double- or three-vessel disease in respectively 20%, 30%, and 40%.101 A severely obstructive lesion containing a variable amount of dynamic thrombus over a plaque is most often identified, providing a rationale for coronary revascularization. On inspection and histologic analyses, the culprit lesion is clearly distinct from the stable plaque which is most often of only moderate severity, with an inner core rich in cholesterol and cholesterol esters and a thin fibrous cap, poor in connective tissue and smooth muscle cells.5 At microscopy, the culprit lesion is rich in monocyte-macrophages, mast cells, lymphocytes, and neutrophils. Biologically, it is extremely active, with an intense inflammatory reaction marked by heterotypic cell-to-cell interactions and activity of proinflammatory cytokines, matrixdegrading metalloproteinases, and growth factors.97 This culprit lesion is the site of a rupture or fissure, occurring most often at the shoulder region of the plaque. The endothelial disruption is overlain by a thrombus extending variably within the lumen of the artery and the vessel wall.5 A number of intravascular imaging technologies have allowed description of the culprit lesion in the living man, including intravascular angioscopy, ultrasound (IVUS), thermography (recording plaque temperature), optical coherence tomography (OCT) (generating images from backscattered reflections of infrared light), intravascular spectroscopy, and magnetic resonance imaging-based approaches.102
Thrombus formation is triggered by tissue factor (TF), which is expressed by lipid-laden macrophages in the core of the atherosclerotic plaque and diseased endothelium. When exposed to the circulating blood, TF binds circulating factor VIIa (FVIIa). The complex triggers the proximal coagulation cascade by activating factor IX and X within the tenase complex. Factor X forms a quaternary complex with TF-FVIIa and tissue factor pathway inhibitor (TFPI) to limit the generation of TF-FVIIa and the thrombogenic stimulus. The thrombus will progress, however, when large enough amounts of FXa are produced. FXa is pivotal in the coagulation cascade. It converts prothrombin to thrombin within the prothrombinase complex. Thrombin has multiple pathophysiologic effects, converting fibrinogen to fibrin, activating factor XIII, which cross-links fibrin, and amplifying its own generation by activation of factors V, VIII, and XI on the platelet surface. It is the most potent platelet agonist in vivo, acting mainly through the thrombin protease-activated receptor type 1 (PAR-1). Additional thrombin receptors in human platelets are PAR-4 and GP Ib-IX-V. Platelet activation is promptly associated with an outside translocation of the inner anionic phospholipid layer of platelets, providing a membrane surface well suited to the assembly of coagulation factors and thrombus formation and growth. Circulating platelets also promptly adhere to the damaged endothelium through receptor–ligand interactions. Gp Ib/IX recognizes von Willebrand factor present in large quantities in the subendothelium, and Gp Ia/IIa recognizes collagen. Platelet adhesion and other local agonists produce intracellular signaling that increases cytosolic Ca2+ content and induces shape change, release of potent vasoactive, proaggregant and procoagulant substances, and activation of Gp IIb/IIIa receptors.5 Activated Gp IIb/IIIa receptors recognize and bind the RGD sequence of various moieties, particularly fibrinogen, resulting in platelet cross-bridging and aggregation. P-selectin, CD40L, and other compounds secreted by activated platelets attract leukocytes, linking mechanisms of thrombosis and inflammation.
The mainstay of immediate therapy in ACS is the control of the thrombotic activity to prevent its rapid progression to occlusion or distal microembolization of thrombotic material. The best results have been achieved with combinations of antiplatelet and anticoagulant therapy consistent because of the contributions to arterial thrombosis of both platelet activation/aggregation and intravascular coagulation.
The goals of treatment in ACS are to control ischemia, decrease the substantial early risk of myocardial infarction and death, and prevent recurrences of the syndrome. These objectives can be collectively regrouped under the terms stabilization or passivation of culprit lesions during the acute phase, and secondary prevention of the underlying atherosclerosis thereafter. They are best achieved acutely by judicious use of anti-ischemic therapy, antithrombotic agents, and reperfusion procedures, then by a program of risk factor control. There are no clear boundaries between these two phases. Treatment is generally guided by risk evaluation based on patient demographics, clinical presentation, the 12-lead ECG, troponin levels, and other patient characteristics associated with an enhanced risk (Table 29.2). Risk stratification is an ongoing process that must be repeatedly updated during the hospital stay and follow-up after discharge.
Table 29.2 Determinants of prognosis of non-ST segment elevation ACS
| Confirming high-risk ACS | Clinical pattern of pain ST-T ischemic changes Troponin T or I elevation Hemodynamic or electrical instability Recurrent ischemia Previous aspirin use |
| Other cardiovascular | Left ventricular dysfunction Congestive heart failure Previous myocardial infarction Previous CABG Extensive coronary or vascular disease Provocative testing |
| Other non-cardiac | Older age Bleeding Diabetes Chronic renal failure Depression |
| Biomarkers | BNP, NTpro-BNP CRP Other markers (e.g. MCP-1, IL-6, MPO) |
Initial measures (generally Class I, Level C)
Patient’s response to symptoms
The patient may first consult with family or colleagues, may visit a medical clinic, doctor’ s office or emergency department, or may call a healthcare provider, optimally 911. Rapid access to the emergency cardiac care system increases the likelihood of prehospital care and prompt prehospital recording of a 12-lead ECG, and rapid transport to an appropriate center for prompt optimal therapy.
Medical triage
Patients are further evaluated in an emergency department where blood levels of biomarkers of infarction are obtained. Patients with milder symptoms such as new onset of angina and/or mild exacerbation of previously stable angina, stable hemodynamics, no angina at rest, no ECG changes and normal serial troponin levels can be further triaged by provocative testing before discharge or as an outpatient soon after discharge. If coronary artery disease (CAD) is suspected, educational material is provided and initial treatment is instituted and re-evaluated at subsequent follow-up. Patients at intermediate risk might go to a coronary care unit (CCU), an intermediate care unit or to a regular ward depending on the availability of facilities and the specific level of risk. High-risk patients should go to the CCU for prompt treatment and are considered for urgent coronary angiography.
General measures
Ischemia at rest or low threshold activity mandates measures to minimize myocardial oxgen demand, including rest in bed or a recliner chair and stool softeners. Emotional distress is minimized by judicious control of the environment, supportive medical and nursing care, limitation and education of visitors, provision for restful sleep, and the control of ischemic pain whenever present with nitrates, intravenous narcotics and other specific anti-ischemic agents as appropriate. Morphine (1–5 mg IV to be repeated if needed) is a useful adjunct, although a note of caution has been raised, by a finding in a large observational registry, of a higher adjusted likelihood of death in patients administered morphine (OR 1.41, 95% CI 1.26–1.57).103 A randomized trial may be warranted. Oxygen is administered to patients with cyanosis or respiratory distress or if finger pulse oximetry reveals an SaO 2 under 90%. Attention is indicated to detect depressive symptoms which may adversely affect prognosis independently of other predictors.53
Anti-ischemic therapy
Nitroglycerin
Nitroglycerin remains a mainstay of angina therapy. It is converted to nitric oxide to cause endothelium-independent vasodilation of capacitance veins (resulting in decreased venous return and preload) and of arterioles (resulting in a reduction in blood pressure and afterload). Together these effects strikingly reduce wall stress and myocardial oxygen demand. Myocardial oxygen delivery may be enhanced if small coronary arteries are concomi-tantly dilated and no coronary steal occurs. The decrease in myocardial oxygen demand is partly offset by reflex increases in heart rate and contractility, which can be counteracted by adequate beta-blockade.
Nitroglycerin (NTG) is given as a sublingual tablet, by oral or sublingual spray or as an intravenous bolus for the immediate relief of chest pain; long-acting oral or transdermal nitrates or nitroglycerin intravenous infusion are useful to prevent recurrent angina, and to rapidly control blood pressure during the acute phase. Intravenous NTG may be initiated at a rate of 10 μ g per min through continuous infusion via non-absorbing tubing and increased by 10μ g increments every 3–5 min until there is some relief of symptoms or blood pressure response, or headache occurs which is not promptly controlled by acetominophen. In general, systolic blood pressure should not be titrated to less than 110 mmHg in previously normotensive patients or to more than 25% below the starting mean arterial blood pressure if hypertension was present.
To minimize the rapid tolerance which occurs with nitroglycerin, a drug-free interval of 6–12 hours a day is generally recommended. This may be undesirable in unstable patients in whom tolerance can be attenuated by reducing the infusion rate once chest pain and blood pressure are under control and increasing the rate subsequently if needed. A ceiling dose of 200 μ g per min is empirically advised although prolonged (2–4 weeks) infusion at rates of 300–400μ g per min has not resulted in increased methe-moglobin levels.104
Nitroglycerin should be avoided in patients with initial systolic blood pressure less than 90 mmHg and in patients with marked bradycardia or tachycardia. Pre-existing hypovolemia is common in the elderly and may predispose to nitroglcerin-induced hypotension. Side effects include headache and hypotension. Hypotension associated with inappropriate bradycardia, reflecting vagal stimulation, may be corrected by Trendelenburg positioning and volume administration or the administration of atropine or glycopyrrolate. Even though the half-life of NTG is short, exacerbation of ischemic changes following abrupt cessation of intravenous NTG has been described105 and a graded reduction in the intravenous dose is advisable.
The phosphodiesterase-5 inhibitors used for the treatment of erectile dysfunction decrease the biodegradation of cyclic guanosine monophosphate (cGMP) whereas organic nitrates also increase levels of cGMP by activating guanylate cyclase. Synergistic decreases of both systolic and diastolic pressure can result in an excessive fall in blood pressure which has been associated with profound hypotension, MI, and even death. The duration of the interaction is variable depending on the half-life of the various drugs. It is advised not to administer nitrates for 4–6 half-lives after the use of a phophodiesterase-5 inhibitor (24 hours for sildenafil and vardenafil and 48 hours for tadala-fil). A recent study of healthy individuals suggested that the sildenafil–nitroglycerin interaction could be gone after four hours.106 The presence of even trace amounts of nitrates could have deleterious effects in combination with a PDE-5 inhibitor, and the administration of sildenafil or another PDE-5 inhibitor to a patient who has taken a nitrate in the preceding 24 hours is contraindicated, as is the administration of any nitrate within 24–48 hours of the administration of a PDE-5 inhibitor. In general, PDE-5 inhibitors should be avoided altogether in patients who require nitroglycerin for treatment of their angina.107 Most studies of nitrates in unstable angina were small, employed case series or case–control designs, and dose regimens varied considerably.108 Many issues such as nitrate tolerance were not addressed. Partial relief of anginal episodes was usually achieved, occasionally relief was complete, and absence of benefit was infrequent. Thus, the widespread use of oral, topical, and IV nitrates in unstable angina is based upon reasonable extrapolation from pathophysiologic observations, case series, evidence of modest reduction of mortality in acute MI,109–111 and extensive clinical experience using regimens developed in careful clinical studies111 (Class I, Level C).
Beta-blockers and calcium antagonists
Based on their effectiveness in the treatment of stable angina, beta-blockers were widely used for the management of unstable angina in the absence of objective evidence for their efficacy. As the calcium channel blockers became available and their effectiveness for the control of stable angina was demonstrated, they also began to be used for the management of unstable angina, and therapeutic trials generally focused on comparisons of beta-blockers and various calcium antagonists. A small randomized and placebo-controlled trial of 126 patients hospitalized with unstable angina showed similar protective effects with nifedipine and the combination of propranolol/isosorbide dinitrate to prevent recurrent chest pain during an evaluation period of 14 days.112 It was noted that the propranolol/isosorbide combination was more effective than nifedipine in patients not taking beta-blockers prior to admission, whereas nifedipine was more effective among patients already on beta-blockade. In the subsequent HINT study,113 338 patients not receiving beta-blocker on admission were randomly allocated, double blind, to nifedipine, metoprolol, both or neither. Metoprolol was significantly more effective than nifedipine (P < 0.05) in preventing acute MI or recurrent angina with ST change. The 177 patients already on a beta-blocker at admission were randomly allocated, double blind, to nife-dipine or placebo, and nifedipine was superior (P > 0.05). In a trial of patients receiving “optimal” doses of nitrates and nifedipine who were then randomized to the addition of either propranolol or placebo,114 propranolol was effective. In another study in patients who had failed maximum treatment with propranolol and long-acting nitrates and were then randomized to the addition of nifedipine or placebo, nifedipine was effective (P > 0.03).115
Diltiazem was compared to propranolol in a randomized single-blind study of patients hospitalized for angina accompanied by ECG abnormalities and occurring in a crescendo pttern, at rest or following MI.116 Chest pain frequency was significantly reduced by both regimens, and there was no difference in efficacy. In another study, patients with rest angina were randomized to diltiazem or propranolol in maximum tolerated doses.117 The agents were equally effective in reducing the frequency of daily anginal episodes, but in the subgroup with angina only at rest, diltiazem was efficacious whereas propranolol was not. The Multicenter Diltiazem Postinfarction Trial Research Group randomly assigned 2466 patients with a non-W wave MI to diltiazem (240 mg daily) or placebo for 12–52 months (mean 25). Mortality rates were nearly identical among the two treatment groups. First recurrent cardiac events (death from cardiac causes or non-fatal rein-farction) were 11% fewer in the diltiazem group (202 vs 226; HR 0.90; 95% CI 0.74–1.08). A significant (P = 0.0042) bidirectional interaction between diltiazem and pulmonary congestion was observed on X-ray examination. In patients without pulmonary congestion, diltiazem was associated with a reduced number of cardiac events (HR 0.77; 95% CI 0.61–0.98); in patients with pulmonary congestion, diltia-zem was associated with an increased number of cardiac events (HR 1.41; 95% CI 1.01–1.96). A similar pattern was observed with respect to the ejection fraction, dichotomized at 0.40.118 A retrospective analysis of the DAVIT trial with verapamil suggested the same detrimental effect on mortality rates in patients with LV dysfunction.119 Small placebo-controlled trials of verapamil demonstrated statistically significant reductions in the frequency of ischemic pain, but longer-term follow-up showed a relatively high incidence of AMI and death.120 Yusuf et al121 examined five trials involving about 4700 patients with threatened MI who were placed on intravenous beta-blocker followed by oral therapy for about a week. There was a modest 13% reduction in the risk of development of MI in this group. Although a meta-analysis of verapamil122 therapy indicated a favorable effect on outcomes, the Yusuf meta-analysis found no overall reduction of death and non-fatal MI with calcium antagonists among patients with unstable angina.121
Altogether these trials suggest that patients not receiving a beta-blocker or a calcium antagonist prehospitalization may benefit from a beta-blocker, diltiazem or verapamil. However, the the meta-analytic data for benefit of beta-blockers but not calcium antagonists and the evidence for improved long-term outcomes with beta-blocker therapy among survivors of myocardial infarction121 and those with chronic ischemia, support beta-blockers over rate-limiting calcium antagonists as the first-choice therapy in patients with unstable angina (Class I, Level A). Patients at high risk may benefit from initial intravenous beta-blocker, followed by an oral regimen (Class I, Level C). Diltiazem or verapamil are suitable alternatives for patients with a contraindication to beta-blocker therapy (Class I, Level C). A short-acting dihydropyridine without a beta-blocker is contraindicated as it can increase the incidence of MI123 (Class III, Level A). A long-acting dose preparation or an agent with an intrinsically long half-life such as amlodipine appears to be preferable, but there has been no rigorous assessment (Class I, Level C). Patients already receiving a beta-blocker can benefit from the addition of a calcium antagonist to prevent recurrent angina (Class I, Level A). Beta-blockers, diltiazem and verapamil should be avoided in patients with severe LV dysfunction (Class III).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


