Antiarrhythmic Drugs: Introduction
Pharmacokinetics of Antiarrhythmic Drugs
Pharmacokinetics describes the relationship between drug administration and plasma concentration. It is assumed that the plasma concentration reflects, in a general sense, myocardial concentrations and thus the availability of the drug at its site of action. The pharmacokinetics of antiarrhythmic drugs are variable and not predictable by drug classification or by knowledge of the drug’s chemical structure.
Absorption describes the movement of orally administered drug from the gut lumen to the systemic circulation, and the efficiency of this absorption is defined as bioavailability. Those factors that influence passage of the drug through the gut wall into the portal circulation and through the liver will have an effect on drug bioavailability. Thus, diseases that affect bowel motility, bowel wall blood flow, gastric and bowel pH, and presystemic or “first-pass” hepatic clearance will, to some extent, influence the amount of drug that is available to the systemic circulation.
A common way to extend interdosing intervals during treatment with drugs that have a relatively short plasma half-life is to formulate them into a sustained-release preparation. Changes in bowel pH and other conditions within the gut influence the amount of drug that is released from sustained-release preparations, which, in turn, has an effect on the amount of drug that is absorbed from the gastrointestinal tract. Drugs can also have altered absorption because of intraluminal binding by other substances, such as milk and antacids. Finally, the physiochemical properties of the drug itself can have an effect on absorption.
Varying the salt of the drug can change solubility and thus its rate of absorption. Drug solubility is also determined by the pH of its medium. Weak bases are rapidly dissolved in acid medium. Antacids, by increasing gut pH, can slow the absorption of a weakly basic drug. In clinical situations, high intestinal pH, caused by such factors as bacterial overgrowth syndrome, can reduce the bioavailability of antiarrhythmic drugs. Thus, for patients with serious arrhythmias, especially those that are controlled by drugs that have a relatively narrow toxic-to-therapeutic ratio, reassessing plasma concentration or clinical efficacy during conditions of altered bowel physiology or pathology is relevant.
Finally, many antiarrhythmic drugs have a food-fast effect; that is, the bioavailability of a drug may be changed several-fold in the presence of food or during fasting. A notable example is oral amiodarone, which is three times more bioavailable after a high-fat meal.1 For such drugs, providing the patient with specific information as to the timing of drug ingestion with relationship to meals is important to maintain a predictable and reproducible plasma concentration over the course of therapy.
Generic antiarrhythmic drugs are approved by demonstration of bioequivalence. Thus, variations in formulation that can occur with generic drugs are permissible as long as there is a clear demonstration that the same amount of the generic drug is absorbed and available to the systemic circulation as the proprietary compound. Regulatory agencies have specified limits in the difference in bioavailability that may be allowed for the approval of new formulations.2
Following absorption, drug is distributed to various parts of the body, and the pattern of distribution is determined by the amount of blood flow to different tissues (Fig. 45–1). Organs with high blood flow, such as the heart, liver, brain, and kidney, are generally exposed to the drug early, and these organs are generally referred to as the central compartment. However, drugs nearly always redistribute from the central compartment to other tissues or sites of metabolism and elimination. They may distribute to muscle, skin, and adipose tissue, generally in a second phase. These less-perfused organs are considered to be the peripheral compartment. Finally, some drugs may distribute very slowly to tissues and organs that have low blood flow and may be referred to as the deep compartment. When the relative concentrations of drugs in all compartments reach equilibrium, steady state has been reached.
Figure 45–1
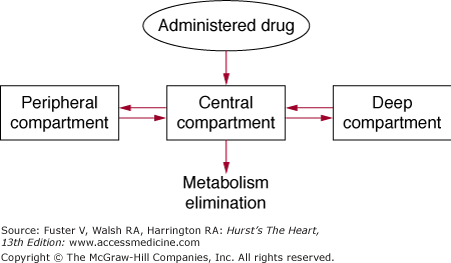
Pharmacokinetic compartments. Administered drug enters a highly perfused central compartment and then is redistributed to peripheral compartments and more slowly equilibrating deep compartments. Reproduced with permission from Siddoway L. Pharmacologic principles of antiarrhythmic. In: Podrid P, Kowey PR, eds. Cardiac Arrhythmias: Mechanisms, Diagnosis and Management. Baltimore, MD: Williams & Wilkins; 1995:359-363.
Volume of distribution is the term used to describe the virtual pool into which a drug is administered. The magnitude of the volume of distribution is dependent on the perfusion of tissues, the concentration of plasma and tissue proteins that bind drug, and the physiochemical properties of the drug as it relates to its binding to tissue and plasma proteins. The volume of distribution is changed in many pathologic conditions, including congestive heart failure. Because organ perfusion is reduced, the volume of distribution of the central compartment is smaller in heart failure, making more of the drug available to active binding sites in the heart. Consequently, patients with congestive heart failure have increased sensitivity to the electrophysiologic properties of commonly used agents.3
Critically important among the factors that modulate volume of distribution is protein binding. Most antiarrhythmic drugs are bound to varying extents in plasma to α1-acid glycoprotein. A variation in protein binding changes the amount of unbound drug that is available to exert a pharmacologic effect. Protein binding is modulated by changes in the concentration of binding proteins, fluctuations in drug concentration, and interactions with drugs competing for binding by the same proteins. The latter is the explanation for drug interactions that occur during simultaneous dosing with drugs such as digoxin and quinidine.4 Changes in plasma and tissue pH can also change protein binding by altering the ionized state of the antiarrhythmic drug. Such alterations are probably only important during marked alterations in pH as may occur during severe acidosis.
The metabolism of drugs in general, as well as of antiarrhythmic drugs, can be divided into two phases. The first phase changes the drug into more polar metabolites via oxidation-reduction reactions. During the second phase, drugs are conjugated to endogenous ligands, such as glucuronide or sulfate. The majority of drug metabolism occurs in the liver, with oxidative metabolism usually occurring via the cytochrome P450 enzyme system. However, the metabolism of rapidly cleared drugs may occur in lung, plasma, vascular endothelium, or even red blood cells.
Knowledge of the relative concentration of active metabolites and their electrophysiologic action is critical to understanding the net pharmacologic effect of an antiarrhythmic drug. An example of an active metabolite is 5-hydroxypropafenone, a metabolite that has similar electrophysiologic effects to those of the parent compound but with slightly less potency.5 The 5-hydroxy metabolite of propafenone possesses little β-adrenergic blocking activity. Thus, the presence of higher concentrations of the metabolite would be associated with less rate slowing in extensive metabolizers compared with a small percentage of individuals who are poor metabolizers of propafenone who manifest a greater β-blocking effect. Similarly, N-acetylprocainamide, the principal metabolite of procainamide, has a greater repolarization effect than the parent drug procainamide. N-acetylprocainamide (NAPA) is renally eliminated, and so in a rapid acetylator with renal impairment, a greater effect on QT interval might be expected compared with an individual who has lesser amounts of the metabolite in the circulation.6
The relative concentration of parent and metabolite in any individual is genetically determined.7 Differences in drug metabolism have been demonstrated for several antiarrhythmic drugs, including propafenone and procainamide. Genetic polymorphisms have been most extensively described for the oxidative P450 enzyme system. Population studies demonstrate that this enzyme system is expressed in a bimodal pattern, with subjects having either “extensive” or “poor” metabolic capability. The poor metabolizer phenotype is expressed in approximately 10% of the population in whom parent drug plasma concentration is increased, clearance is reduced, and elimination half-life is prolonged. P450 enzyme isoforms have been identified that are responsible for the metabolism of specific agents and now can be identified by genotyping. Careful monitoring of patients during drug initiation to detect differences in response that may be caused by relative concentration of parent and metabolite is critically important for patient safety.
CYP2D6 is the enzyme responsible for the metabolism of propafenone, flecainide, acebutolol, metoprolol, and propranolol. Most patients are extensive metabolizers, which means that a large percentage of the parent compound is transformed into a metabolite with activity that is identical to, similar to, or dissimilar from the parent. CYP3A4 is the most prevalent enzyme in the liver and metabolizes many antiarrhythmic drugs, including quinidine, lidocaine, mexiletine, and many calcium channel blockers. This enzyme is inhibited by cimetidine, erythromycin, and grapefruit juice. Use of these agents causes accumulation of higher concentrations of the parent compound and thus an exaggerated electrophysiologic effect.
Metabolism issues are important in predicting the interaction of antiarrhythmic drugs with other agents, including other cardiac and noncardiac drugs that may be metabolized by the same pathways. These drugs may interfere with metabolism either by occupying the enzyme site or by having a direct effect on the activity of the enzyme. The relative affinity of the drug for the enzyme system is predictive, in many cases, of the potency of the drug interaction that might occur with commonly used agents such as digoxin and warfarin.7
Drug elimination occurs via a number of routes in addition to hepatic metabolism. Clearance is the term that describes removal of drug from plasma and is defined as the volume of plasma cleared of drug per unit of time. The sum of all individual clearances is called total-body clearance and is the most reliable indicator of the rate of metabolism and elimination of drug. Other methods of quantifying elimination are less satisfactory. Terms such as half-life can be altered both by changes in clearance and by volume of distribution.
Clearance generally begins with presystemic clearance, which is the metabolism of the drug during its passage through the liver before reaching the systemic circulation, as previously described. A number of drugs, such as lidocaine,8 undergo extensive first-pass clearance, which obviates their oral use. Other drugs with less extensive first-pass clearance may be given at high doses so as to make some of the drug available to the systemic circulation. Examples include propafenone, propranolol, and verapamil. Although most drugs have a linear relationship between oral dose and plasma concentration during first-pass metabolism, others such as propafenone exhibit saturable kinetics, resulting in a nonlinear increase in plasma concentration during increases in oral dosing. This also results in a change in the ratio of parent drug to metabolite when the oxidative enzyme in the liver becomes saturated.
Despite its limitations, the concept of half-life can be used to describe the time required for drug elimination. Half-life is defined as the time required for the plasma concentration to decrease by 50% (Fig. 45–2). For drugs that have more than one pharmacologic compartment, more than one half-life may be defined. For example, the decrease in plasma concentration after administration of an intravenous bolus of a drug can be divided into its distribution period, when the drug is distributed from the central to the peripheral compartments, and an elimination period, during which time the drug is cleared from the central compartment by metabolism and elimination. Conversely when a drug is administered by mouth, it is usually absorbed at a rate slower than the rate of distribution of the drug. Therefore, the distribution phase may be masked by the slow absorption of the drug.
Figure 45–2
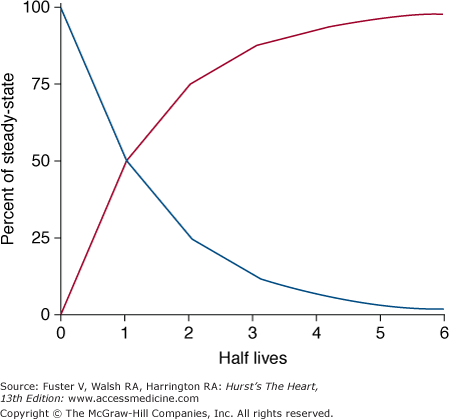
Time required for accumulation and elimination of drug. After initiation of drug therapy (red line), 50% of steady-state drug concentrations are achieved in 1 half-life and 90% are achieved in 3.3 half-lives. Similarly, after drug is stopped (blue line), concentration decreases by 90% after 3.3 half-lives. Reproduced with permission from Siddoway L. Pharmacologic principles of antiarrhythmic. In: Podrid P, Kowey PR, eds. Cardiac Arrhythmias: Mechanisms, Diagnosis and Management. Baltimore, MD: Williams & Wilkins; 1995:359-363.
As can be seen from Fig. 45–2, the time required to achieve a steady-state plasma concentration during chronic therapy can be estimated from the elimination half-life. After approximately 3.3 half-lives, 90% of the steady-state plasma concentration has been reached during loading or removed during washout. Thus for a drug with a half-life of 8 hours, a period of 24 hours is sufficient to reach near steady-state levels.
For drugs that have a longer half-life and therefore a longer time to achieve steady state, a loading period may be used to bring plasma concentrations into a desired range more rapidly. Lidocaine is an excellent example of this principle. Using fixed infusion rates without loading would require >6 hours for lidocaine to achieve a steady-state plasma concentration. In the setting of treatment of patients with severe arrhythmias, this time period is unacceptably long. Therefore, bolus doses are given to fill the central compartment and to replace the amount of drug leaving the plasma through distribution to peripheral compartments.8 Once steady state has been achieved—that is, when there is no more movement of drug from central to peripheral compartments—the drug can be administered as a maintenance infusion that is given at the same rate at which the drug is eliminated.
Amiodarone represents the most complicated example of drug loading to balance distribution and elimination.9 Amiodarone is generally considered to have three compartments, all of which need to be saturated to achieve steady state. Large doses of oral drug are given over several weeks to achieve saturation of the peripheral and deep compartments. However, because the myocardial half-life of the drug is significantly shorter than the half-life of deeper compartments, the drug is given daily to maintain equilibrium in the myocardium.
Knowledge of the elimination half-life is critical for designing proper interdosing intervals for most oral antiarrhythmic drugs. The main considerations in determining how frequently a drug has to be administered are half-life and therapeutic range, defined as the minimum and maximum plasma concentrations that will achieve clinical benefit without exposing patients to toxicity.10 The larger the ratio of maximum to minimum therapeutic effect, the longer may be the intradosing interval, during which time high peak and low trough concentrations will be achieved. Reducing peaks and valleys requires shorter interdosing intervals. For most drugs, knowledge of renal elimination kinetics is critical in formulating an effective interdosing interval and finding an adequate dose to achieve a therapeutic effect. A notable example of this principle is sotalol. Sotalol is entirely eliminated through the kidney. An estimate of creatinine clearance is necessary to select an appropriate dose and dosing interval.
One caveat in designing dosing schedules is the problem of active metabolites. Metabolites may have pharmacokinetics that are distinct from the parent.5,6 For drugs that have metabolites that are active and have longer half-lives than the parent, interdosing intervals might need to be extended to account for the metabolite’s effect. Accumulation of a metabolite with higher than necessary dosing frequency can produce toxicity that may be life threatening.
Finally, drug half-life may be determined not only by the rate but also by the route of elimination. A prime example is adenosine, which is rapidly taken up and metabolized by erythrocytes and vascular endothelium and thus eliminated in prompt fashion.11 Drugs with rapid offset of effect frequently are eliminated by routes other than the kidney, or they may be distributed quickly from the central compartment as in the case of ibutilide.12
The elderly are particularly prone to overdosing with antiarrhythmic drugs. There are several physiologic changes associated with aging that make drug accumulation more likely. These include reduction in lean body mass, decrease in renal function, alterations in hepatic metabolism, and changes in receptor affinity.13 The elderly also have increases in glycoprotein concentrations and reductions in serum albumin concentrations that may alter the ability of drugs to bind to plasma proteins, in effect raising free concentrations. Reduced receptor affinity and stimulation of adenylate cyclase attenuate the effects of catecholamines and may make these patients more susceptible to the depressant effects of β-adrenergic blocking agents. Reduction in the activity of the renin-aldosterone system and arterial wall elasticity make elderly patients more susceptible to the development of orthostatic hypotension. Although it is clear that aging also affects the density of ion channels, no age-related changes in the physiology of the ion channel per se has been documented. Because of these physiologic changes, elderly patients in general should be started on a smaller dose of an antiarrhythmic drug with slow upward dose titration to clinical effect or to tolerance.
In general, patients with larger body size require higher doses of drugs to achieve a therapeutic plasma concentration. However, drugs may distribute differently in adipose tissue compared with muscle and other tissue. Thus, if adjustment in dose is made only on the basis of the patient’s weight, some drugs will be overdosed.
The central volume of distribution of an antiarrhythmic drug is generally reduced in congestive heart failure, necessitating reduction in initial dose.14 Congestive heart failure also gives rise to derangements of liver and renal blood flow with reduction in clearance of the drug and high extraction ratios. For all of these reasons, as well as altered sensitivity to drug and altered plasma protein binding, drugs should be administered in low dose to patients with congestive heart failure with slow upward dose titration, as in the elderly.
Drug and metabolites can accumulate in patients with renal failure, depending on the proportion of the drug that is eliminated by the kidney. Drugs such as sotalol, which are exclusively renally eliminated, are particularly important in this regard.15 Fortunately, most drugs have other routes of elimination, which can at least partly compensate for reduced elimination because of renal failure. Amiodarone is an example of a drug that is not at all eliminated by the kidney and that can be safely administered to patients on renal dialysis and to patients who are functionally anephric.
Fortunately, until hepatic disease is far advanced, hepatic metabolism and elimination of most drugs is not significantly compromised. Short of liver cirrhosis, it can be assumed the drugs will be metabolized and eliminated normally, even in patients who have elevated transaminase and bilirubin concentrations.10
Antiarrhythmic drug interactions are ubiquitous and, unfortunately, underappreciated by practicing physicians. This is a complex issue because drug interactions may occur for a number of reasons, including inhibition or enhancement of drug metabolism and elimination, displacement of drug from plasma or tissue-binding sites, competition for receptor sites, and additive pharmacodynamic effects.10 Table 45–1 outlines common drug interactions. Interaction of drugs at the level of the P450 enzyme system is responsible for an appreciable number of drug interactions in clinical practice. Other important drug interactions occur because of interference with elimination, such as when digoxin clearance is reduced by agents such as propafenone, flecainide, and amiodarone. The mechanism of this interaction has been described best for quinidine, which also dislodges digoxin from tissue-binding sites, leading to potentially toxic digoxin levels. Among the assorted other mechanisms for kinetic interactions is reduced hepatic blood flow by β-adrenergic blockers, which can result in reduction of the clearance of high extraction drugs such as lidocaine.
Mechanisms Underlying Antiarrhythmic and Proarrhythmic Effects
The development of cardiac arrhythmias and drug-induced proarrhythmias can be a result of abnormal impulse formation (ie, enhanced automaticity and triggered activities) and/or reentry (see Chap. 38). This section provides a brief overview of ionic and cellular mechanisms underlying antiarrhythmic and proarrhythmic effects of the drugs.
Antiarrhythmic drugs exert their effects on cardiac electrical impulse formation and propagation via their interaction with ionic channels or with membrane receptors and cellular pumps that subsequently influence ionic currents across the cell membrane. Cardiac ionic currents commonly targeted by antiarrhythmic drugs include inward sodium current (INa), L-type calcium current (ICa,L), and delayed rectifier outward potassium current (IK), which consists of two components—a rapidly activating component (IKr) and a slowly activating component (IKs). Although transient outward potassium current (Ito) is not commonly influenced by currently available antiarrhythmic drugs, it has been recently shown to contribute importantly to the genesis of polymorphic ventricular tachycardia and ventricular fibrillation.16 Therefore, antiarrhythmic drugs with selective Ito blockade will be favored in future drug development.
Antiarrhythmic drugs that block the sodium channel appear to bind selectively to the channel during only one or two of the states and dissociate from the channel during the other states (Fig. 45–3). Therefore, a sodium channel blocker will block the sodium channel differently depending on pathologic conditions. Because the maximal velocity of change in membrane potential during phase 0 (Vmax) is correlated with influx of sodium ions through opened sodium channels and the major driving force for electrical conduction in atrial, His-Purkinje, and ventricular tissues, sodium channel blockers that bind to the channel during the open state, like class Ia and Ic drugs, cause conduction slowing that manifests as P wave and QRS widening on the body surface electrocardiogram (ECG). However, sodium channel blockers that predominantly bind to the channel in the inactivated state, like class Ib drugs, have only a modest effect on action potential phase 0 under normal condition. In ischemic myocardium with reduced membrane potential, voltage- and time-dependent recovery of sodium channel from inactivation is delayed, so that binding of class Ib drugs to the channel is significantly increased. This explains the greater activity of class Ib antiarrhythmic drugs under conditions of myocardial ischemia. In contrast, atrial repolarization is faster compared with the ventricles and is associated with rapid transition of the atrial sodium channel from the inactivated to the resting state. Therefore, class Ib drugs play little role in atrial arrhythmias. A few channels may remain open through sustained depolarization, the so-called late (or slow) INa17 and contribute to the action potential duration. Compared with the epicardium and endocardium, the submyocardium of the left ventricle (M cells) has a larger late INa.18 Blockade of the late INa causes action potential shortening, whereas an increase in this current by drugs like ibutilide leads to action potential lengthening.
Figure 45–3

Schematic of sodium channel gating. Depolarization of the cell membrane opens the m gate (activation) and initiates the process (ie, inactivation) for closing the h gate at the same time, so that the sodium channel remains open only transiently (1-2 ms) before cycling into the inactivated state. Recovery of the sodium channel from the inactivated state for reopening is voltage and time dependent.
ICa,L is responsible for the prolonged plateau phase (phase 2) of the action potentials of atrial, His-Purkinje, and ventricular cells and is the major inward current responsible for phase 0 depolarization in sinus and atrioventricular (AV) nodal cells. Consequently, calcium channel blockade causes slight action potential shortening in the atria, His-Purkinje system, and ventricles; depresses the slope of diastolic depolarization and action potential amplitude; and prolongs the effective refractory period in sinus and AV nodal cells. L-type calcium channels may reactivate during the action potential plateau under conditions of delayed ventricular repolarization (ie, long QT syndrome). This may lead to early afterdepolarization (EAD) capable of initiating a specific form of polymorphic ventricular tachycardia termed torsade de pointes (TdP).19 In addition, larger calcium influx via L-type calcium channel increases the likelihood of calcium overloading in the sarcoplasmic reticulum (SR) under certain pathologic conditions. Oscillatory release of calcium from SR increases Na/Ca exchange, resulting in the genesis of delayed afterdepolarization (DAD) and triggered activity.20
IKr and IKs are the major repolarizing currents in the heart, and blockade of them leads to an increase in action potential duration and effective refractory period in the atria, AV node, His-Purkinje fibers, ventricles, and accessory pathways. Therefore, the drugs that inhibit IKr and/or IKs, such as sotalol, dofetilide, and azimilide, have value in the treatment of reentrant arrhythmias like atrial fibrillation, atrial flutter, and monomorphic ventricular tachycardia. The majority of antiarrhythmic drugs that prolong QT interval and induce TdP block IKr.
The role of Ito in arrhythmogenesis has been highlighted in the past one and half decades. A prominent Ito-mediated action potential notch in the ventricular epicardium, but not endocardium, produces a transmural voltage gradient during early ventricular repolarization that could register as a J wave or J point elevation on the ECG.21 If the Ito-mediated notch is deep enough, epicardial action potential dome at phase 2 is sensitive to a change in the net repolarization current. An increase in the net repolarization current, due to either a decrease in inward currents or augmentation of outward currents, may result in partial or complete loss of the dome, leading to a transmural voltage gradient that manifests as ST-segment elevation.22 Heterogeneous loss of Ito-mediated action potential dome, particularly when it is complete, results in marked repolarization dispersion on the epicardium surface, leading to reentry at action potential phase 2 (ie, so-called phase 2 reentry). The extrasystole produced via phase 2 reentry occurs often on the down limb of the T wave (R-on-T phenomenon). This may, in turn, initiate polymorphic ventricular tachycardia or fibrillation.22 In structurally normal hearts, the Ito-mediated J wave is the intrinsic link for ST-segment elevation among early repolarization syndrome, idiopathic ventricular fibrillation, and Brugada syndrome. Therefore, these syndromes are now referred to as J wave syndromes.16 This explains why quinidine, a class IA drug that also exhibits an additional inhibitory effect on Ito, is effective in preventing ventricular fibrillations in J wave syndromes.23
Blockade of inward currents may be use dependent. The class Ic drugs dissociate from the sodium channels in the resting state very slowly, exhibiting strong use dependence that manifests as a marked increase in sodium channel blockade during tachycardia. This is thought to be responsible for the increased efficacy of the class Ic antiarrhythmic drugs in slowing and converting tachycardia with minimal effects at normal sinus rates. However, strong use dependence of a drug on INa may be proarrhythmic (eg, flecainide-induced atrial flutter or ventricular tachycardia) when the effect of the drug on conduction slowing is stronger than its effect on the effective refractory period.
As opposed to the use dependence observed for drugs that block the sodium and calcium channels, inhibition of IKr is enhanced during bradycardia when the channel is “not frequently used,” leading to more significant action potential prolongation at slow heart rates. The mechanism is probably related to the dependence of IKr blockade on extracellular potassium concentration.24 Reverse use-dependent properties of antiarrhythmic drugs may reduce their efficacy in tachycardias and predispose to bradycardia-dependent proarrhythmias such as TdP.
Development of reentry using an anatomic circuit, for example around a scar tissue secondary to myocardial infarction, is determined by the wavelength of the reentrant wavefront and the size of the circuit. The wavelength of the reentrant wavefront is equal to the product of the conduction velocity times the effective refractory period of myocardial tissue in the pathway and should be significantly shorter than the length of the reentrant circuit for maintenance of circus movement. Conduction velocity of the electrical impulse in the atria, His-Purkinje, and ventricles is largely determined by the intensity of the fast inward sodium current that contributes to the rapid upstroke (Vmax) of the action potential. For example, class Ic antiarrhythmic drugs may predispose to the development of monomorphic ventricular tachycardia in patients with coronary artery disease when the drugs’ effects to reduce Vmax of the action potential and conduction velocity surpass their effects to prolong the effective refractory period.25 However, antiarrhythmic drugs that prolong the effective refractory period without influencing conduction and thus the wavelength of a reentrant impulse are useful in suppressing reentrant arrhythmias.
Normal automaticity of cardiac cells is the consequence of spontaneous diastolic depolarization caused by a net inward current during phase 4 of action potential. Abnormal automaticity, which is caused by either enhanced normal automaticity or spontaneous activity in ventricular and atrial myocardium, may occur with heightened β-adrenergic tone or reduced resting membrane potentials, such as ischemia or infarction.26 β-adrenergic receptor or calcium channel blockers are useful in treatment of arrhythmias due to abnormal automaticity. The hyperpolarization-activated “funny” current (If; also called pacemaker current) may play an important role in enhanced automaticity such as in inappropriate sinus tachycardia. Blockade of this current is a promising treatment option for patients with inappropriate sinus tachycardia.27
Afterdepolarization can be divided into two subclasses: EADs and DADs. EADs often occur in M cells or endocardium because these action potentials prolong disproportionally with drugs that block IKr, an ionic current targeted by most QT-prolonging drugs.19,28 Antiarrhythmic drugs that inhibit ICa,L or shorten action potential duration by blocking the sodium current, such as mexiletine, suppress EADs. Calcium channel and β-adrenergic blockers suppress DADs and, therefore, DAD-mediated arrhythmias by reducing intracellular calcium overload.
Proarrhythmic and antiarrhythmic effects of a drug are two sides of the same coin. Antiarrhythmic drugs that are intended to suppress arrhythmias may potentially worsen a preexisting arrhythmia or cause a new arrhythmia. The mechanisms responsible for cardiac arrhythmias are complicated, and any intervention may be antiarrhythmic in some circumstances and proarrhythmic in others.
When class Ic antiarrhythmic drugs are used for the treatment of supraventricular tachycardia, they can cause atrial flutter, often with slower rates (~200 beats/min). This is seen in patients with atrial fibrillation treated with the class Ic drugs flecainide and propafenone. The slowed atrial flutter may be associated with 1:1 AV conduction with markedly rapid ventricular rates. Therefore, dosing with an AV nodal blocking agent should be considered with a sodium channel blocker in patients with atrial fibrillation and intact AV nodes.
Adenosine and digitalis occasionally promote the development of atrial fibrillation by shortening atrial effective refractory period. Shortening of the refractory period reduces the wavelength of reentrant wavefront, so that reentry can occur in smaller reentrant substrates.
Ventricular proarrhythmias can generally be divided into two categories based on mechanisms and ECG features: monomorphic ventricular tachycardia and a specific form of polymorphic ventricular tachycardia termed TdP.
A key factor in the genesis of monomorphic ventricular tachycardia is the availability of an anatomic reentrant pathway such as scar tissue or tricuspid and mitral rings. Any drug that causes conduction slowing greater than effective refractory period prolongation has the potential to facilitate the development of reentrant arrhythmias. Class Ic drugs have the highest risk to cause this type of proarrhythmia and to increase mortality in patients with coronary artery disease and left ventricular systolic dysfunction.29-31 Myocardial ischemia seems to play a pivotal role in the genesis of proarrhythmia with class Ic drugs. Conversely, proarrhythmia rates are nearly zero when class Ic drugs are used for atrial arrhythmias in patients with a structurally normal heart.32
TdP requires QT prolongation.22 An example is “quinidine syncope” in patients who have taken quinidine for the treatment of atrial fibrillation and experienced recurrent syncope or cardiac arrest as the result of QT prolongation and TdP. It is widely accepted that TdP is initiated by EAD-dependent triggered activity and maintains itself via functional reentry.19 Class Ia and class III antiarrhythmic drugs that block IKr and/or increase late INa may facilitate the development of TdP. Interestingly, amiodarone, a commonly used class III antiarrhythmic drug, significantly prolongs the QT interval but rarely causes TdP. Amiodarone prolongs the action potential duration without producing EADs and reduces transmural dispersion of repolarization, probably as a consequence of its effects on multiple ionic channels and receptors.33 Dronedarone, a noniodinated benzofuran derivative of amiodarone that was approved for treatment of atrial fibrillation in 2009, also causes QT prolongation without a significant risk of TdP.34
Marked QT prolongation and resultant TdP are more likely to occur in patients with reduced repolarization reserve. Clinical diseases or factors that are associated with reduced repolarization reserve include congenital long QT syndrome, bradycardia, female sex, ventricular hypertrophy, electrolyte disturbances such as hypokalemia and hypomagnesemia, and coadministration of other QT-prolonging agents or drugs that delay clearance of the drugs from the body.
A number of antiarrhythmic drugs have significant hemodynamic effects that may limit their usefulness under certain clinical situations. It is well known that class I drugs reduce myocardial contractility and may exacerbate congestive heart failure.35 Among the sodium channel blockers, class Ic drugs and disopyramide exert the most potent negative inotropic effect; β-adrenergic and calcium channel blockers also are negative inotropic agents because of their inhibitory effect on inward L-type calcium current. Although β-adrenergic blockers, when used in patients with compensated heart failure, reduce symptoms of heart failure and improve survival, their use in patients with decompensated heart failure who need inotropic support of intravenous agents should be avoided.
The majority of class III antiarrhythmic drugs are well tolerated hemodynamically in patients with heart failure. Data from clinical trials show that dofetilide and amiodarone do not significantly affect the hemodynamic state of patients with severe left ventricular dysfunction and heart failure.36,37 However, d,l–sotalol should be used cautiously in patients with significant heart failure because its β-adrenergic blocking effect may exacerbate heart failure.
The antiarrhythmic drug classification system most often used was originally put forth by Vaughn-Williams and modified by Harrison. This classification is relatively simple and useful. It assumes that each antiarrhythmic drug has a predominant electrophysiologic mechanism of action and has a primary therapeutic application (Table 45–2).
| Class | Action | Example Drugs |
|---|---|---|
| I | Sodium channel blockade | |
| Ia | Block sodium channel in open state with an intermediate recovery from block; also, inhibit IKr at relatively lower concentrations; moderate phase 0 depression and conduction slowing, prolonging of action potential duration | Quinidine, procainamide, disopyramide |
| Ib | Block sodium channel in inactivated state with a fast time constant of recovery from block; minimal effect on phase 0 upstroke; no change or slight shortening of action potential duration | Lidocaine, mexiletine |
| Ic | Block sodium channel in open state with slow recovery from block; marked phase 0 depression and conduction slowing; small or no effect on repolarization | Flecainide, propafenone |
| II | β-Adrenergic receptor blockade | Propranolol, metoprolol, atenolol, esmolol, acebutolol, pindolol, nadolol, carvedilol, labetalol and bisoprolol |
| III | Potassium channel blockade and/or inward current enhancer | d,l–Sotalol, dofetilide, amiodarone, bretylium, ibutilide, dronedarone |
| IV | Calcium channel blockade | Verapamil, diltiazem |
According to the original schema, drugs that have an effect on sodium channels were placed in class I. It was later recognized that the relative potency of all of these drugs varied and that some had additional electrophysiologic effects differentiating them from others (see Table 45–2
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


