Type
Fibril protein precursor
Clinical syndrome
AA
Serum amyloid A protein
Reactive systemic amyloidosis associated with chronic inflammatory diseases
AL
Monoclonal immunoglobulin light chains
Systemic amyloidosis associated with monoclonal plasma cell dyscrasias
AH
Monoclonal immunoglobulin heavy chains
Systemic amyloidosis associated with monoclonal plasma cell dyscrasias
Aβ2M
β2-microglobulin
Periarticular and, occasionally, systemic amyloidosis associated with long-term dialysis
ATTR
Normal plasma transthyretin
Senile systemic amyloidosis with prominent cardiac involvement
ATTR
Genetically variant transthyretin
Autosomal dominant systemic amyloidosis
Familial amyloid polyneuropathy
ACys
Genetically variant cystatin C
Hereditary cerebral haemorrhage with cerebral and systemic amyloidosis
AGel
Genetically variant gelsolin
Autosomal dominant systemic amyloidosis
Predominant cranial nerve involvement with lattice corneal dystrophy
ALys
Genetically variant lysozyme
Autosomal dominant systemic amyloidosis
Non-neuropathic with prominent visceral involvement
AApoAI
Genetically variant apolipoprotein AI
Autosomal dominant systemic amyloidosis
Predominantly non-neuropathic with prominent viscera involvement
AApoAII
Genetically variant apolipoprotein AII
Autosomal dominant systemic amyloidosis
Non-neuropathic with prominent renal involvement
AFib
Genetically variant fibrinogen A alpha chain
Autosomal dominant systemic amyloidosis
Non-neuropathic with prominent renal involvement
ALect 2
Leukocyte chemotactic factor 2
Slowly progressive renal amyloid with nephrotic syndrome and liver involvement
It is interesting to note that given the right conditions nearly any polypeptide chain can be driven towards misfolding and aggregation but relatively few proteins are amyloidogenic in vivo [2]. The ultrastructural morphology and histochemical properties of all amyloid fibrils, regardless of the precursor protein type, are remarkably similar [3–5]. During amyloidogenesis, multimeric proteins dissociate to their monomeric components, and may further be enzymatically cleaved before or during their conversion into amyloid fibrils [6, 7]. There are essentially three circumstances in which amyloid deposition occurs. The first is when there is sustained abnormally high abundance of proteins that are normally present at low levels, such as serum amyloid A protein (SAA) in chronic inflammation, underlying susceptibility to AA amyloidosis. The second is when there is normal abundance of a normal, but to some extent inherently amyloidogenic protein over a very prolonged period, such as transthyretin in senile amyloidosis (ATTRwt). The third situation is the presence of an abnormal protein with markedly amyloidogenic structure, such as certain monoclonal immunoglobulin light chains in AL amyloidosis and genetic variants of transthyretin, apolipoprotein AI and fibrinogen Aα chain etc in the autosomal dominant diseases of hereditary amyloidosis.
Despite the heterogeneity of the various precursor proteins, the morphological structure and histochemical properties of all amyloid fibrils are remarkably similar. Diffraction studies of amyloid fibrils have confirmed that they all share a common core structure consisting of anti-parallel β-strands lying perpendicular to the long axis of the fibril [8, 9]. This extremely abnormal highly ordered conformation underlies the distinctive physicochemical properties of amyloid fibrils. The fibrils are relatively stable and are resistant to proteolysis. All amyloid fibrils possess the ability to bind molecules of the dye Congo red in a spatially organised manner which results in the pathognomonic apple green birefringence when viewed under cross polarised light. Amyloid deposits also always contain the normal plasma glycoprotein, serum amyloid P component (SAP) as a non-fibrillar constituent. The universal presence of SAP in amyloid deposits [10] reflects its specific binding to an, as yet, uncharacterised ligand common to all amyloid fibrils which forms the basis for diagnostic scintigraphic imaging of amyloid with radiolablelled-SAP [11].
The clinical phenotype of amyloid deposition is remarkably diverse, ranging from an asymptomatic small, localised deposit to a systemic, rapidly lethal multisystem disease [12]. Almost any organ can succumb to the effects of fibril deposition including the lung and in most instances the disease presents with multi-organ involvement. While amyloid deposits may be widespread clinically important amyloidosis results from the disruption of the structure, integrity and function of affected tissues and organs. The natural history of amyloidosis is usually of progressive accumulation. Although there is continual low-grade turnover of amyloid deposits, clinical progression reflects the fact that the fibrils are being laid down faster than they are cleared away. Amyloid deposits can therefore regress if the balance is tipped in favour of clearance. As such, many of the currently available treatment strategies have focussed on reducing the supply of fibrils by halting the production of the culpable plasma protein. The primary goal of therapy is thus to prevent further progressive amyloid deposition. Although most types of amyloidosis are progressive and unremitting, there are numerous reports describing actual regression of amyloid when the underlying inflammatory or other condition have been successfully treated [13–19]. Current efforts are being directed at developing treatment strategies which may result in more pronounced disappearance of the amyloid fibrils although human trials have not yet been completed in this regard [20].
Diagnosis and Evaluation of Amyloidosis
As amyloidosis is a remarkably heterogenous disease it may present to a variety of different medical specialties. There are numerous reasons for patients with amyloidosis to present to the respiratory physician. Chronic pulmonary conditions can themselves give rise to systemic amyloidosis, most commonly of AA sub-type. While these patients rarely present with symptomatic involvement of the lung the underlying pulmonary disease is the driver of the amyloidogenic protein production and it is therefore important to recognise those patients at risk. Patients with systemic amyloidosis may also present with respiratory symptoms as a consequence of amyloidosis itself whereby amyloid deposits are found in the lung as a component of a more systemic process. Localised, isolated pulmonary and respiratory tract amyloid deposits are also well-described and may either present with symptoms or be detected incidentally on chest radiography or biopsy. Lastly, it is important to recognize that, especially in the context of AL amyloidosis, pulmonary complications may also arise from treatment.
The key first step in diagnosis is obtaining tissue evidence of amyloid fibril deposition. The diagnostic gold-standard for the presence of amyloid is histological confirmation by Congo red staining, producing red-green birefringence under crossed polarised light [3] (Fig. 7.1a, c). Most tissue specimens, ranging from needle biopsies to open surgical resections, can be studied although small biopsies are open to sampling error given the often focal nature of the deposits. Standard formalin fixation of the biopsy specimen is appropriate in the majority of cases. Samples can then be embedded in paraffin for cutting. These samples are suitable for the Congo red stain as well as subsequent immunofixation. It is also appropriate for laser capture microdisection and mass spectrometry described later in the chapter. Biopsy of any organ can be hazardous in amyloidosis as there is an increased risk of haemorrhage. Significant bleeds having been reported in 5 % of liver biopsies [21], although more recent data from renal biopsies is reassuring [22]. A case report from 1987 described fatal lung haemorrhage following transbronchial biopsy in a patient with amyloidosis. The post mortem findings showed the biopsied blood vessels infiltrated with amyloid [23]. A further case series by Utz et al. published in 1996 reported no major complications in 11 patients following transbronchial lung biopsies however 2 of the 11 cases were reported to have 100 mL blood loss [24]. A less invasive alternative in suspected disease is fine needle aspiration and this has been used successfully in the respiratory tract [25, 26]. The increased risk of haemorrhage in amyloidosis is multifactorial. There is often increased fragility in involved blood vessels as well as reduced elasticity of amyloidotic tissues. Very occasionally in AL type amyloid an acquired deficiency of clotting factors IX or X is seen [27–29]. Interestingly the factor deficiencies are most often due to sequestering of the circulating factor in the liver and spleen. It is not generally due to deficient factor production or the development of an inhibitor as is sometimes seen in other clonal lymphoproliferative disorders.
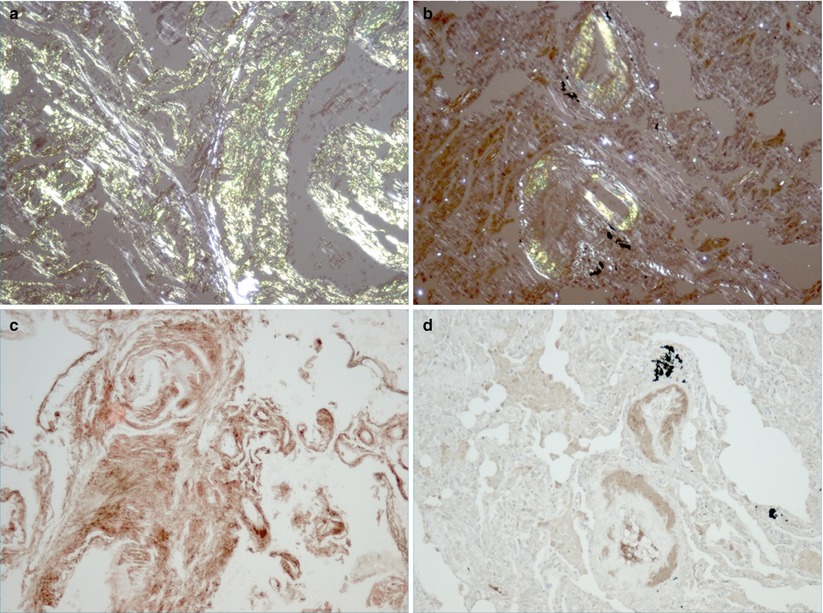
Fig. 7.1
Tissue confirmation of amyloid subtypes. Lung biopsies showing characteristic histological appearance of amorphous amyloid deposits stained with Congo red from patients with lambda light chain restricted AL amyloid (a) and ATTRwt amyloid (b). Confirmatory testing is shown by immunohistochemistry using anti-lambda light chain antibodies (c) and anti-transthyretin antibodies (d)
Once amyloid is identified immunohistochemical stains are then used to determine the fibril protein type as this ultimately guides further therapeutic interventions (Fig. 7.1b, d) [30]. Suitable antibodies for most of the common amyloid subtypes are widely available. This includes antibodies to kappa and lambda light chains, SAA and transthyretin. It should be noted that although immunohistochemistry is usually definitive in AA amyloidosis, it is non-diagnostic in about 20 % of AL deposits [31, 32]. Expertise in the immunohistochemical typing of hereditary amyloid is restricted, and definitive immunohistochemical typing of amyloid deposits cannot always be achieved [33]. Laser capture micro-dissection and mass spectrometry is fast being established as the new gold-standard in fibril typing. The technique involves micro-dissection of a suspected amyloid deposit, identified by positive Congo red staining, from a cut specimen on a microscope slide. The microscopic protein deposit is removed from the slide and then analyzed by mass spectrometry to identify the fibril sub-type, this technique is successful in the vast majority of cases. It is especially useful in those patients who cannot be confidently diagnosed through immunohistochemical typing however the technique is currently limited by availability [34]. If Congo red positivity is not demonstrated but the clinical suspicion of a clonal lymphoproliferative disorder being the source of the symptoms is still high one should consider Nonamyloidotic Monoclonal Immunoglobulin Deposition Disease (NMIDD). This may be due to either heavy chain or light chain deposition within the tissues but without amyloid fibril formation. This is a rare entity but the presentation can be very similar to more typical AL amyloid of the lung. The diagnosis is again dependent on tissue biopsy often requiring special pathology expertise. Clinically, management is very similar to that of AL amyloid given that the underlying etiology is a clonal disorder.
If a genetic variant is suspected one should pursue more detailed analyses examining for mutations in the gene giving rise to the amyloidogenic fibril. In general sequencing is the preferred modality and ideally samples should be sent to a reference lab with expertise in this area. A web-based repository reviewing all currently known mutations in genes with amyloidogenic potential has been recently made available to help guide these investigations (http://amyloidosismutations.com).
Once histological confirmation of amyloid has been obtained the extent of the organ deposition and dysfunction needs to be established. In respiratory tract amyloidosis this can be challenging and the optimum imaging technique can vary depending upon the distribution of deposits. In many cases however, it may be normal. Plain radiography as an initial assessment can be helpful with findings ranging from local amyloidomas presenting as intrathoracic mass lesions (Fig. 7.2a), to non-specific reticular interstitial infiltrates (Fig. 7.2b). Computed tomography (CT) scanning is particularly useful in further defining interstitial disease (Fig. 7.2c). In combination with positron emission tomography (PET) imaging it can also help to better define the metabolic activity of a solid lesion thus aiding differentiation from more typical intrathoracic malignancy or metastases as well as rare entities such as plasmacytomas [35]. In addition magnetic resonance imaging (MRI) and bronchoscopy may also be useful in combination with comprehensive pulmonary function tests (PFTs). PFTs are an important objective tool to formally establish the severity of clinically relevant disease and are useful in guiding therapeutic decisions. Evidence of extra-pulmonary systemic disease should be sought clinically and by performing relevant serologic and radiographic investigations. A diagnostic algorithm is presented in Fig. 7.3.
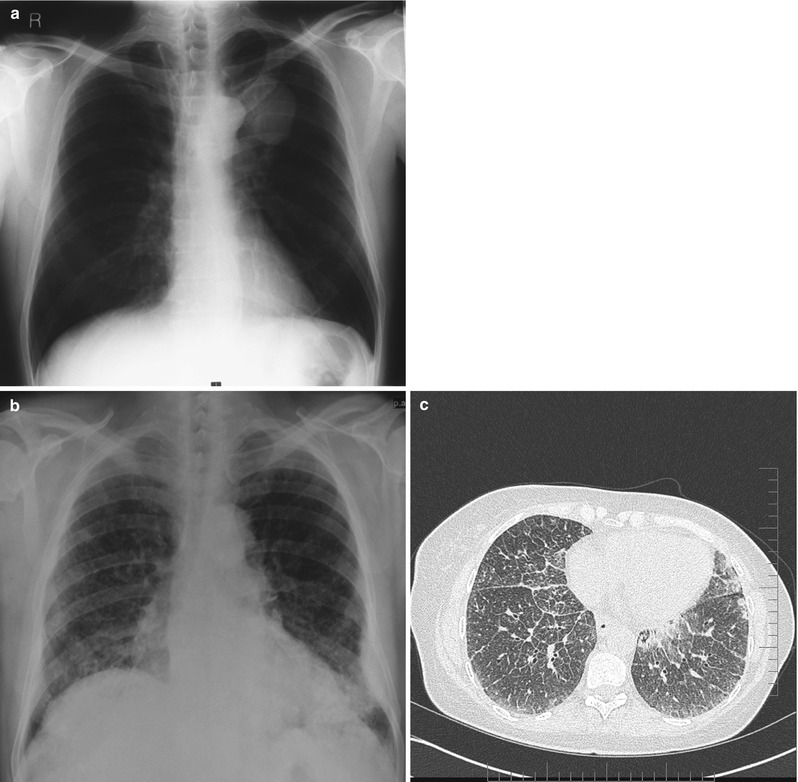
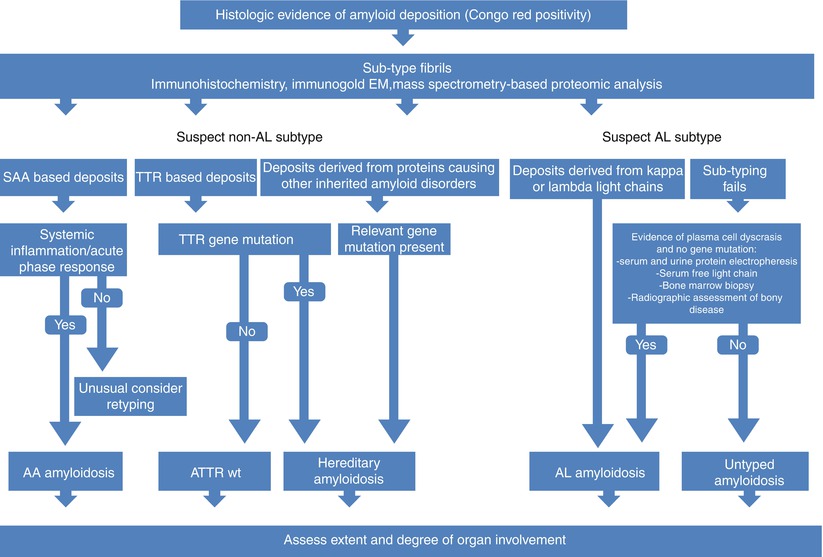
Fig. 7.2
Radiologic appearance of amyloid deposition. Chest x-ray showing isolated pulmonary amyloidoma (a). Chest X-ray demonstrating the typical intersitioal pattern associated with parenchymal amyloid fibril deposition (b). High resolution CT of the chest on the same patient confirming intersitioal infiltration (c)
Fig. 7.3
Algorithm for investigations of patients with suspected amyloidosis
If AL amyloid is suspected it is important to identify the underlying clonal disease process. The plasma cell clones that underlie systemic AL amyloidosis are often subtle and may not be detected by bone marrow examination or immunofixation of serum and urine. In recent years, the use of the serum free light chain assay has increased the diagnostic sensitivity in testing for clonal disease [36, 37]. Immunoglobulin gene rearrangement studies may identify subtle clones in either the bone marrow or, in the case of localised AL, within the amyloidotic tissue itself [38]. A number of other non-invasive techniques are available to assess the overall amyloid burden. Radiolabelled 123I-human SAP localises specifically to amyloid deposits in vivo in proportion to the quantity of amyloid present and thereby enables diagnosis, quantification and monitoring of amyloid [11]. SAP scintigraphy is useful in visualising amyloid in the major organs of the abdomen (Fig. 7.4a). Localisation to the lungs is poor thus it is not routinely used for identifying or monitoring amyloid deposits in the respiratory tract however dense fibril deposits as seen in large amyloidomas may occasionally show distinct tracer uptake (Fig. 7.4b). In combination with single photon emission computed tomography (SPECT)-CT imaging SAP uptake can be better delineated; better characterizing the degree of uptake within an organ or lesion (Fig. 7.4c). It is also important to note that while it is a very specific test for the presence of amyloid, alone it cannot reliably differentiate the fibril sub-type. Cardiac amyloidosis is best evaluated by a combination of echocardiography and ECG [39–41]. Two-dimensional Doppler echocardiography classically reveals concentric biventricular wall thickening with a restrictive filling pattern [42]. Amyloid primarily causes diastolic dysfunction with a decline in the ejection fraction occurring generally in the latter stage of the disease [40]. The ECG may be normal but in advanced disease commonly shows small voltages, pathological ‘Q’ waves (pseudo-infarct pattern) in the anterior chest leads and conduction abnormalities. Cardiac magnetic resonance imaging (CMR) is being used more frequently and is extremely useful in identifying cardiac amyloid. Typical appearances are of homogenous late gadolinium enhancement [43]. 99mTc-3, 3-diphosphono-1,2-propanodicarboxylic acid (99mTc-DPD) scintigraphy is fast becoming a powerful imaging tool in the investigation of cardiac amyloid. It is a specific test indicative of fibril deposition within the heart with newly emerging data suggesting that the degree of uptake and pattern of distribution in the extra-cardiac soft tissue may even be specific for ATTR amyloid [44–46]. If these studies are confirmed prospectively then this may evolve as a tool whereby a non-invasive diagnostic test can be used to confirm fibril sub-type. Elevation of N terminal Pro brain naturetic peptide (NT-Pro BNP) and cardiac troponins can also be helpful in establishing whether a patient has cardiac amyloid. Together they have become key markers to risk stratify patients and guide therapy [47]. It is important to recognize that these enzymes are not always specific and can be elevated for many reasons such as renal impairment and other forms of cardiomyopathy; however a normal NT-Pro BNP can often exclude clinically significant cardiac involvement [48, 49].
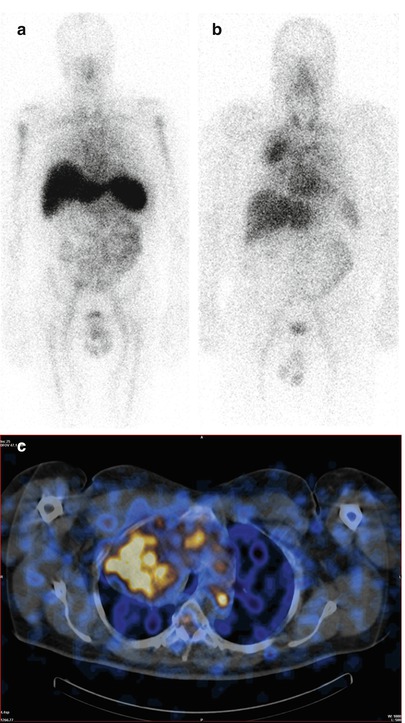
Fig. 7.4
Scintigraphic assessments using 123I-human SAP. An anterior whole body scintigraphic image from a patient obtained following intravenous injection of 123I-human SAP showing abnormal uptake into the amyloid deposits within the spleen, liver and bone marrow (a). An anterior plasmacytoma and deposition in the spleen. An anterior whole body scintigraphic image from a patient with a solitary intrathoracic amyloidoma (b) with corresponding SPECT-CT (c)
Systemic Amyloidosis Complicating Respiratory Diseases
Systemic AA Amyloidosis
Systemic AA amyloidosis is a potential complication of any disorder associated with a sustained acute phase response. The list of chronic inflammatory, infective or neoplastic disorders that can underlie it is almost without limit (Table 7.2). Biopsy and post-mortem series suggest that the prevalence of AA amyloid deposition in patients with chronic inflammatory diseases is between 3.6 and 17 %, though a smaller proportion of patients have clinically significant amyloidosis [50, 51]. The amyloid fibrils are derived from cleavage fragments of the circulating acute phase reactant, SAA [52]. SAA is an apolipoprotein of high density lipoprotein (HDL) [53], which is synthesised by hepatocytes under the transcriptional regulation of cytokines including IL-6, IL-1 and TNF-α [54]. In health the circulating concentration of SAA is around 3 mg/L, but this can rise by more than a thousand fold in the presence of inflammation. The circulating concentration of SAA tends to parallel that of the much more frequently measured C-reactive protein (CRP). A sustained high plasma level of SAA is a prerequisite for the development of AA amyloidosis but why amyloidosis develops in only a small proportion of cases remains unclear. AA amyloidosis can present anytime between childhood and old age with a median age at presentation of 48 years in the UK. It is slightly more common in men and, although disease can develop very rapidly, the median latency between presentation with a chronic inflammatory disorder and clinically significant amyloidosis is almost two decades [55].
Table 7.2
Conditions with Respiratory Manifestations associated with systemic AA amyloidosis
Chronic infections | Neoplasia |
Bronchiectasis | Adenocarcinoma of the lung, |
Leprosy | Carcinoid tumour |
Q fever | Castleman’s disease |
Subacute bacterial endocarditis | Hodgkin’s disease |
Tuberculosis | Mesothelioma |
Immunodeficiency states | Inflammatory arthritis |
Common variable immunodeficiency | Adult Still’s disease |
Cyclic neutropenia | Ankylosing spondilitis |
Hyperimmunoglobulin M syndrome | Juvenile idiopathic arthritis |
Hypogammaglobulinaemia | Rheumatoid arthritis |
Sex linked agammaglobulinaemia | Systemic vasculitis |
HIV/AIDS | Behcet’s disease |
Other conditions predisposing to chronic infections | Systemic lupus erythematosis |
Cystic fibrosis | Other |
Kartagener’s syndrome | SAPHO syndrome |
Paraplegia | Sarciodosis |
Sickle cell anaemia | Sinus histiocytosis with massive lymphadenopathy |
The most common respiratory disease underlying AA amyloidosis in the United Kingdom is bronchiectasis. It is the fifth commonest cause, accounting for 5 % cases. A study of 16 patients with end stage renal failure secondary to AA amyloidosis due to bronchiectasis in Turkey, reported the mean duration of bronchiectasis to be 22.2 years, with a wide range ± 12.2 years. The mean age at presentation was 50.6 ± 13.5 years. Eight cases (50 %) had cystic bronchiectasis, four of whom died from suppurative pulmonary infections. The other eight patients had chronic fibrotic changes, four of these eight cases were considered to be the sequelae of previous TB infections [56]. It is important to recognize however that the prevalence of bronchiectasis is falling due to both earlier treatment of necrotizing pneumonia and prevention of pulmonary infections via routine immunization programmes.
Neoplasia including primary lung malignancies, lymphoma, adenocarcinoma and polyclonal lymphoid diseases like Castlemans disease account for approximately 3 % of AA amyloid cases [57–61]. Castleman’s disease (angiofollicular lymph node hyperplasia) is a rare B cell lymphoproliferative disorder characterized by giant hyperplastic lymph node follicles, capillary proliferation and plasma cell infiltration [62, 63]. It is often associated with a significant systemic inflammation and marked constitutional symptoms. It comprises solitary and multicentric forms, and there are hyaline vascular and plasma cell variants histologically [64]. Multicentric disease, commonly of the plasma cell type, usually has an aggressive and rapidly fatal course. It is most often seen in the context of HIV infection and thought to be related to human-herpes virus 8 infection with the disease driven by either host or virus derived IL-6. In Asia there is also a well described entity of multicentric Castleman’s not associated with HHV-8 but still driven by cytokine overproduction [63]. Unicentric disease tends to occur in younger patients and is of the hyaline vascular type in more than 70 % of cases, and of plasma cell type or mixed histology in the remainder [65]. Most solitary tumours occur within the mediastinum and consist of a dominant mass surrounded by multiple enlarged lymph nodes which, histologically, may appear merely reactive. Constitutional symptoms including night sweats, fever and weight loss are common and laboratory abnormalities including anaemia, elevation of the erythrocyte sedimentation rate (ESR) and polyclonal hypergammaglobulinaemia are almost universal. Acquired systemic amyloidosis is a recognized rare complication of all forms of angiofollicular lymph node hyperplasia, and is usually of systemic AA type occurring as a result of the persistent cytokine-driven acute phase response [58]. Surgical resection of unicentric tumours can result in complete remission and excellent long term outcome [58, 66]. Chemotherapy, anti-IL6 immunotherapy and anti-virals have also been explored in the treatment of this condition, tocilizumab (humanized monoclonal anti-IL6 receptor antibody) is now established as the treatment of choice in this disease [67–69]. While in Castleman’s disease IL-6 seems to be the primary driving factor of systemic inflammation in other lymphomas the exact pathophysiology driving the systemic inflammatory response is not well understood. However, it is again hypothesized that tumour derived cytokines are produced which stimulate the synthesis of serum amyloid A protein by the liver [70].
Other purely respiratory causes of AA amyloidosis are now fairly rare in the UK although in the first half of the last century tuberculosis was the commonest single disease resulting in AA amyloid. This remains the case in many parts of the developing world and is likely to be vastly underreported. Other less common underlying conditions include cystic fibrosis, sarcoidosis and Kartagener syndrome.
In general, AA amyloidosis is a systemic condition that results in organ dysfunction due to widespread deposition of AA amyloid fibrils. The primary and often the first organ involved is the kidney, usually presenting with proteinuria. Progressive renal dysfunction follows often accompanied by overt nephrotic syndrome. Splenic amyloid deposits are almost universally present and are detectable by SAP scintigraphy. This is often of little clinical significance and patients are usually entirely asymptomatic with the amyloidotic spleen frequently not palpable. Hepatic involvement and autonomic neuropathy are well recognised but usually occur only very late in the disease. Cardiac AA amyloidosis is extremely rare occurring in less than 2 % of cases often as a late finding. Overt respiratory tract involvement has not been a clinical feature among over 400 patients with systemic AA amyloidosis evaluated in our own unit. In systemic amyloidosis it is not uncommon to see small deposits of amyloid in the blood vessels of most tissues including the lungs, however these deposits are usually asymptomatic and regarded as incidental. Although there have been a few reports of systemic AA amyloidosis affecting the lungs, fibril typing was generally imperfect [71]. All studies in which the fibril protein has actually been sequenced identified AL type. As emphasized previously diagnosis of amyloid relies on a high index of clinical suspicion and requires histological confirmation.
SAP scintigraphy provides a useful non-invasive method of diagnosis at the UK National Amyloidosis Centre. When considering AA amyloid it can be used as a screening examination with greater than 98 % accuracy as the vast majority of these patients will have at least splenic uptake [55]. That said the most effective form of basic screening in medical or respiratory practice is to target patients at risk of developing AA amyloidosis, identifying those with ongoing poorly controlled inflammation and to performing urinalysis at each clinic attendance. More than 95 % of patients with AA amyloidosis will have significant proteinuria on dipstick testing which should prompt further specialist investigation [55]. The prognosis of AA amyloidosis depends on the degree of renal dysfunction at presentation, the chronicity of the inflammatory process and whether the underlying chronic inflammatory disease can be effectively suppressed, so that the plasma SAA is maintained below 10 mg/l. When the supply of fibril precursor protein is substantially reduced by such methods, AA amyloid deposits frequently regress and renal function can improve [55, 72]. If the acute phase response continues unabated, progressive amyloid deposition often results in end stage renal failure. In individuals who present with advanced renal disease even complete suppression of their inflammatory disease may not be sufficient to preserve their renal function and in all cases renal deterioration is accelerated by hypertension. Treatment depends on the underlying diagnosis and may include surgery for cytokine secreting tumours or localised bronchiectasis, long term antimicrobials and postural drainage for chronic infections associated with structural lung problems (as in cystic fibrosis or Kartagener syndrome) and immunosuppression in inflammatory diseases such as sarcoidosis.
Almost 40 % of patients with AA amyloidosis eventually develop dialysis dependent renal failure. Renal outcomes on dialysis are equivalent to that of age matched non-diabetic patients on the end stage program with a median survival of 53 months. Mortality is higher in the first year and this has been attributed to ongoing heavy urinary protein losses and increased risk of sepsis. Nephrotic syndrome is a major risk factor for sepsis, particularly in patients who are predisposed to infection such as those with bronchiectasis. A minority of patients go on to receive renal transplants. The published outcomes are rather variable but our series of almost 40 highly selected patients had excellent outcomes with an 82 % 5-year graft survival [73]. It is recommended in patients with AA amyloidosis that renal transplantation is considered in those who have excellent control of their SAA levels and regular monthly monitoring is recommended to ascertain suitability. Unfortunately patients with underlying respiratory conditions causing AA amyloidosis may be unable to have renal transplants as unresolved bacterial infections and incurable malignant disease remain contraindications.
Systemic AL Amyloidosis
Systemic AL amyloidosis is the commonest type of systemic amyloidosis accounting for approximately 60 % of cases. It may occur in association with any form of monoclonal B cell dyscrasia [74, 75]. The precursor proteins are monoclonal immunoglobulin light chains and generally consist of the whole or part of the variable (VL) domain [76, 77].
A number of conditions localised to the thoracic cavity can underlie systemic AL amyloidosis. Although rare, an isolated plasmacytoma can secrete enough monoclonal free immunoglobulin light chains into the circulation to produce systemic AL amyloid deposits [78]. Rarely, this can present as a chest mass (Fig. 7.4b, c). While more commonly associated with AA amyloid, Castleman’s tumours, both unicentric and multicentric, can be associated with monoclonal immunoglobulin light chain production and are a rare cause of AL amyloidosis [79]. The commonest condition managed by respiratory physicians which can cause both systemic and respiratory localised AL amyloid deposits is Sjogren’s syndrome which is discussed later in the chapter.
Systemic AL amyloidosis is most commonly associated with a clonal proliferation of plasma cells. A degree of amyloid deposition is seen in up to 15 % of patients with overt myeloma, but the vast majority of patients (more than 80 %) who present with clinically significant AL amyloidosis have a low grade plasma cell dyscrasia [80]. AL amyloidosis usually presents in patients over the age of 50 years, although it can present in very young adults [80]. Clinical manifestations are extremely variable since almost any organ other than the brain can be directly involved [81]. Although certain clinical features are strongly suggestive of AL amyloidosis (Table 7.3) and multi-organ dysfunction is common, many patients initially present with non-specific symptoms such as malaise and weight loss. The outlook of untreated AL amyloid is far worse than AA type, with a 5 year survival of approximately 10 % and a 10 year survival of less than 5 % [80]. Most affected individuals eventually die of heart failure, uraemia or autonomic failure. Criteria have been published to formally document involvement of the major organ systems include the lung in AL amyloidosis (Table 7.4).
Table 7.3
Clinical features associated with systemic AL amyloidosis
Organ involvement | Clinical manifestation |
|---|---|
Soft tissue infiltration | Bruising – especially periorbital; Macroglossia; Muscle/joint pseudohypertrophy |
Renal | Proteinuria; Nephrotic syndrome; Nephrotic Syndrome; Hypertension very rarely |
Cardiac | Restrictive cardiomyopathy; Arrhythmias; Congestive cardiac failure |
Pulmonary | Shortness of breath; Restrictive or obstructive defects; decreased diffusion capacity |
Hepatic | Hepatomegaly; Liver failure very rarely |
Peripheral nervous system | Carpal tunnel syndrome; Symmetrical sensorimotor neuropathy |
Autonomic nervous system | Orthostatic hypotension; Impotence; Disturbed bowel motility; Impaired bladder emptying |
Gastrointestinal | Weight loss; Blood loss; Disturbed bowel motility |
Lymphoretiicular | Splenomegaly; Lymphadenopathy |
Adrenal axis | Hypoadrenalism (rare) |
Table 7.4
Criteria defining organ involvement with in AL amyloidosis
Organ | Defining criteria |
|---|---|
Kidney | 24-h urine protein >0.5 g/day, predominantly albumin |
Heart | Echo: mean wall thickness >12 mm, no other cardiac cause |
Liver | Total liver span >15 cm in the absence of heart failure or alkaline phosphatise >1.5 times institutional upper limit of normal |
Nerve | Peripheral: clinical; symmetric lower extremity sensorimotor peripheral neuropathy |
Autonomic: gastric-emptying disorder, pseudo-obstruction, voiding dysfunction not related to direct organ infiltration | |
Gastrointestinal tract | Direct biopsy verification with symptoms |
Lung | Direct biopsy verification with symptoms |
Interstitial radiographic pattern | |
Soft tissue | Tongue enlargement |
Arthropathy | |
Claudication (presumed vascular amyloid) | |
Skin lesions | |
Myopathy (by biopsy or pseudohypertrophy) | |
Lymph node (may be localized) | |
Carpal tunnel syndrome |
The breadth of symptoms associated with pulmonary involvement can be either directly related to respiratory tract amyloid deposits or due to sequelae from extra-pulmonary organ dysfunction. Although microscopic deposits of amyloid are almost universally present in the lungs, in the vast majority of cases dyspnoea is secondary to cardiac involvement. A restrictive cardiomyopathy is found in up to one third of all patients with AL amyloid, ultimately being the cause of death in one half [82]. Renal involvement is also frequent in AL amyloidosis and presents in the same manner as renal AA amyloid [83]. Gut involvement can cause motility disturbances (often secondary to autonomic neuropathy), malabsorption, perforation, haemorrhage, or obstruction [84]. Peripheral neuropathy occurs in one fifth of cases and typically presents with a painful sensory polyneuropathy followed later by motor deficits [80]. Autonomic neuropathy causing orthostatic hypotension, impotence, and gastrointestinal disturbances may occur in isolation or with a peripheral neuropathy [81].
As previously stated the chest radiograph is usually normal in systemic AL amyloidosis. Nodular “amyloidomas” arising from a small local population of clonal plasma cells secreting amyloidogeninc light chain may be detected appearing as focal masses. A diffuse reticulonodular pattern may also be present if the lung parenchyma is involved. Lung function tests sometimes show a restrictive pattern and rarely reduced gas transfer due to extensive alveolar deposits [85]. Persistent pleural effusions have been described in 5.5 % of patients yet this is more commonly associated with amyloid heart disease [86]. Nephrotic syndrome secondary to renal amyloid deposits can also result in gross salt and water overload and often, in combination with cardiac involvement, can result in pulmonary oedema and pleural effusions. Amyloid deposits can occasionally directly involve the pleura causing disruption of normal pleural function and resultant effusions. Chronic pleural effusions secondary to pleural amyloid are often refractory to management with diuretics and require recurrent drainage or pleurodesis [86].
Amyloid Deposits in the Respiratory Tract
First described by Lesser in 1877, localised amyloidosis of the respiratory tract ranges from asymptomatic pulmonary nodules to diffuse parenchymal deposits. Presenting symptoms can mimic a variety of lung pathology and depends on the location of the amyloid fibrils. Initial investigations with routine imaging can be unhelpful in confirming the diagnosis. A number of classifications have been proposed based upon radiographic or bronchoscopic findings but have not been widely adopted [24, 87]. In general amyloidosis is best classified by the fibril protein (Table 7.1), and then by the sites of clinical involvement.
Localised amyloid deposition is not uncommon although often goes undiagnosed. It results either from local production of fibril precursors [88, 89], or from properties inherent to the particular microenvironment, which favour fibril formation from a widely distributed precursor protein [90]. The vast majority of localised amyloid deposits are of AL type [91, 92] with symptomatic deposits occurring most frequently in the eye [93], skin [94], respiratory [24, 95] or urogenital tracts [96, 97]. They are often associated with extremely subtle focal monoclonal B cell proliferation confined to the affected site. Surgical resection of these localised ‘amyloidomas’ can sometimes be curative if the underlying clonal are removed [97]. Symptomatic apparently localised amyloid deposits can rarely be manifestations of systemic disease and patients should always be fully investigated to exclude more generalised amyloid deposition [30].
Thorough evaluation of respiratory tract amyloidosis and biopsy confirmation is required to determine the need for treatment and the most suitable modality based on fibril sub-type. However, the paucity of controlled clinical trials means that management decisions have to be made on an individual basis. Broadly speaking, systemic chemotherapy is usually indicated for systemic AL amyloidosis and local intervention, according to symptoms for its localised forms.
Laryngeal Amyloidosis
The larynx is the most frequent site of localised amyloidosis affecting the head and neck [98]. It represents 0.5–1 % of benign laryngeal disease, its incidence increases with age but it occasionally affects young adults [99]. Discrete nodular and diffuse infiltrative types of laryngeal amyloid were initially described in 1949 [100], the diffuse pattern with an intact mucosa being more common, sometimes with tracheobronchial extension [92]. The macroscopic appearance is often seen as diffuse subepithelial oedema without mucosa or nodular alterations [101]. The amyloid deposits occur most commonly in the ventricles followed by the subglottis, the aryepiglottic folds and the true vocal cords [92]. Presentation is usually with hoarseness or rarely stridor, but can cause a sensation of ‘fullness’ in the throat, choking, and dyspnoea on exertion [102]. The aetiology remains unclear, there is no reported association with alcohol use, smoking, vocal abuse or infections [98]. One proposed explanation for the predilection of the larynx is that the production of light chains may be arising from B-cell clones within mucosal associated lymphoid tissue [95, 103]. Deposits are predominantly lambda in origin [96, 104].
The diagnosis is usually made following larnygoscopy and biopsy. The extent of infiltration is best determined with MRI examining specifically for tracheobronchial extension. On MRI imaging laryngeal amyloid is reported to give intermittent T1-weighted signal intensity and low T2-weighted signal intensity similar to skeletal muscle. It is felt to be superior to CT images when evaluating amyloid of the pharynx, larynx and trachea [105]. Systemic amyloidosis should be excluded and investigations for an underlying plasma cell dyscrasia are imperative. There are case reports of extramedullary plasmacytoma with amyloid deposition affecting the larynx and it is important to distinguish this from a localised deposit of amyloid as this clearly changes the prognosis and approach to management [106].
Very rarely apparently localised laryngeal amyloid deposits can be due to a feature of hereditary systemic apolipoprotein AI amyloidosis (AApoAI). Apolipoprotein AI is a major constituent of high density lipoprotein (HDL) [107]. Wild type apolipoprotein AI is amyloidogenic and is present as traces of amyloid in human aortic atherosclerotic plaques in 10–20 % of autopsies although rarely leads to overtly symptomatic disease [108]. Fourteen separate apolipoprotein variants have been reported to cause this [33, 89, 109, 110] and in three of these, the major site of organ damage is the heart. Numerous other amyloidogenic variants have been reported and are associated with specific genetic alterations and a different pattern of organ distribution. Depending on the mutation, patients can present with massive abdominal visceral amyloid involvement [111], predominant cardiomyopathy [109] or a familial amyloid polyneuropathy like syndrome [112]. Laryngeal and cutaneous deposits producing hoarseness, infiltrative plaques, and petechial rashes are associated with the apolipoprotein AI Arg173Pro, Ala175Pro, Leu90Pro, and Leu178His variants. Case reports suggest that the macroscopic appearances of the laryngeal deposits in AApoAI amyloidosis are different from those seen in localised AL disease, they appear as small irregular floppy proliferations affecting the borders of the vocal folds in contrast to firm bulky deposits in the localised AL form [113].
AApo AI amyloid is autosomal dominantly inherited with variable penetrance and a family history of the disease may therefore be lacking. Performing immunohistochemistry on biopsy specimens for both AL kappa and lambda and apolipoprotein AI is recommended. Genetic sequencing looking for Apo AI mutations should also be performed.
Tracheobronchial Amyloidosis
Tracheobronchial amyloidosis is an uncommon diagnosis although it too may well be underreported. It is characterised by amyloid deposits primarily in the trachea and large bronchi, with extension at times into segmental bronchi and frequent involvement of the submucosal vessels [24, 114]. A literature review from 1983 identified 67 cases of which 57 were diffusely infiltrative (multifocal submucosal plaques) and the remainder were nodular or ‘tumour-like’ [87].
Presenting symptoms include dyspnoea, persistent cough and haemoptysis [115]. Narrowing of airways can cause an obstructive airflow defect and cases of tracheobronchial amyloidosis simulating asthma have been reported. Deposits may also cause distal atelectasis, recurrent pneumonia or lobar collapse and solitary nodules may be mistaken for endobronchial neoplasia [116]. Routine chest x-ray may be misleading. In a recent review of 64 cases, 70 % of cases had normal radiographic findings [117]. Imaging with computed tomography (CT) commonly shows tracheobronchial thickening, stenosis, atelectasis and patchy shadows with no specific findings attributable to amyloid deposition. Magnetic resonance imaging (MRI) may be more helpful in demonstrating more specific features suggestive of amyloidosis. Typically deposits have intermediate T1 weighted signal intensity and low T2 weighted signal intensity similar to skeletal muscle [105]. Dual phase FDG PET/CT imaging can be used to differentiate between malignancy and amyloid deposits. Early phase FDG metabolic activity can be seen but delayed images show reduced activity which would not be seen with malignancy [118]. Given the non-specific nature of the imaging of tracheobronchial amyloidosis the diagnosis is often delayed as it most often requires bronchoscopy and biopsy [119]. Tracheobronchopathia osteoplastica, characterised by calcified or cartilaginous submucosal nodules within the airways [120–122] and relapsing polychondritis are the principle differential diagnoses [123, 124]. Ding et al. reviewed the literature in 2010 and reported the natural history of 64 cases. The median age at diagnosis was 49 (range 21–82) years. The median time to diagnosis was 37 months, with a male to female ratio of 1.21 [117]. Although symptomatic tracheobronchial amyloidosis is usually localised, its course is not always benign and overall survival is only 31–43 % at 4–6 years: three of seven cases followed up by Hui died of respiratory failure or secondary pneumonia [125], and three of four Mayo Clinic patients died within 79 months of diagnosis [24].
Parenchymal Pulmonary Amyloidosis
Amyloid within the lung parenchymal tissue is the most frequently detected respiratory manifestation of amyloidosis [126]. It can be divided radiographically into solitary/multiple nodules or a diffuse alveolar-septal pattern [127, 128].
Nodular pulmonary amyloidosis is almost always due to localised AL deposits and is usually an incidental finding on chest radiography. Although the lesions may appear dramatic and need to be differentiated from neoplasia the prognosis is usually excellent. In theory CT/PET should be useful in distinguishing between amyloid nodules and malignancy. A recent case report however, suggests that PET imaging can give false positive results in nodular pulmonary amyloidosis and thus although it may be a helpful investigation it does not replace the need for a histological diagnosis [129, 130]. Amyloid nodules in the lung parenchyma are usually peripheral and subpleural, occurring preferentially in the lower lobes; they may be bilateral and range in diameter from 0.4 to 15 cm. They grow slowly and may cavitate or calcify [126, 127, 131]. Larger nodules can occasionally produce space occupying effects but otherwise no treatment is required.
Rarely pulmonary amyloid nodules have been reported to be transthyretin amyloid in type (ATTR). Wild type transthyretin amyloidosis is also known as senile systemic amyloidosis and usually presents with cardiac involvement although amyloidogenic transthyretin gene mutations are also well described [114]. Pulmonary involvement in senile systemic amyloidosis seems to be a rare incidental finding at autopsy [114]. A case report described by Roden et al., describes an 82 year old patient who presented with a diffuse nodular infiltrate and recurrent pleural effusions. Echocardiographic findings were consistent with cardiac amyloid. Biopsy of one of the lung tissue confirmed transthyretin amyloid by both immunohistochemistry and mass spectrometry. The TTR gene was found to be wild type by DNA sequencing [132]. Pulmonary nodules associated with AA amyloidosis have been found in patients with rheumatoid arthritis [133], Crohn’s disease [134] and in patients with AA amyloidosis secondary to intravenous drug abuse [71] all have run a reportedly benign course.
Diffuse amyloid deposition within the lung parenchyma is usually associated with systemic AL amyloidosis (Fig. 7.6) with concomitant involvement of other organ systems [114]. Post-mortem series have confirmed that diffuse parenchymal amyloid is common in systemic AL amyloidosis although is symptomatic in a minority of cases [135, 136]. When pulmonary manifestations develop the disease is serious and responds poorly to therapy. Patients are often very symptomatic frequently presenting with dysponea which may be further compounded by amyloid induced cardiac dysfunction. The pathophysiology is due to deposition of the amyloid fibrils within the small airways and the capillary alveolar membrane leading to impaired gas exchange and respiratory failure detectable by decreased carbon monoxide diffusion capacity. With more widespread involvement a restrictive defect can also occur similar to that seen in pulmonary fibrosis. Pulmonary function testing provides a quantitative means of both assessing a patient’s baseline dysfunction and tracking the progression or response to treatment.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


