Adult Congenital Heart Disease
Advances in the treatment of congenital heart disease over the past five decades have led to significant growth in the number of surviving patients. Accurate statistics are lacking, but estimates of adult patients with congenital heart disease in the United States in the year 2000 were more than 750,000 (1). Most adult cases consist of simple defects such as bicuspid aortic valve, right aortic arch, and atrial septal defect. However, patients with more severe forms of congenital disease, such as pulmonary atresia, Ebstein anomaly, and transposition of the great arteries, can also survive into adulthood. For this reason, radiologists should have a broad understanding of the anatomic and physiologic aspects of congenital heart disease. In this chapter we provide a broad overview of adult congenital heart disease, with an emphasis on chest radiography, computed tomography (CT), and magnetic resonance imaging (MRI). The discussion focuses on patients who have not had prior surgical corrective procedures, cases where radiologists can be the first to provide clues for early diagnosis.
The chapter is divided into two sections (Table 19.1). The first section is a discussion of congenital defects that do not produce cyanosis, such as anomalies of the aorta, left-to-right intracardiac shunts, and other miscellaneous conditions. The second section addresses the more complex cyanotic defects.
Noncyanotic Congenital Heart Disease
Bicuspid Aortic Valve
The congenitally bicuspid aortic valve, after mitral valve prolapse, is the second most common major cardiac malformation (2). The malformation occurs as frequently as 2 in every 100 births (3). The finding sometimes remains clinically silent throughout life, found incidentally at autopsy. However, the tendency is to have progressive thickening and fibrosis of the valve with aging. Valve stenosis is the most common complication of this malformation. When stenosis occurs, virtually all adult patients will have calcification of the valve found at pathologic tissue examination. Abundant aortic valve calcification found on chest radiographs in a patient under the age of 50 should be mentioned in reports as a possible diagnosis of valve stenosis, especially if associated with dilatation of
the ascending aortic outline (Fig. 19.1). Incidental identification of abundant aortic valve calcification on chest CT in patients under the age of 55 should also be reported as a possible case of aortic valve stenosis (Fig. 19.2) (4). Congenital subaortic and supraaortic valve stenoses are uncommon conditions in adults shown on occasion on imaging studies (Fig. 19.3).
the ascending aortic outline (Fig. 19.1). Incidental identification of abundant aortic valve calcification on chest CT in patients under the age of 55 should also be reported as a possible case of aortic valve stenosis (Fig. 19.2) (4). Congenital subaortic and supraaortic valve stenoses are uncommon conditions in adults shown on occasion on imaging studies (Fig. 19.3).
Bicuspid aortic valve is the second most common major cardiac malformation after mitral valve prolapse.
Table 19.1: Outline of Congenital Heart Disease | ||||
|---|---|---|---|---|
|
Aortic valve calcification, particularly in individuals less than 50 years of age, has a high association with aortic valve stenosis.
Table 19.2: Common Anomalies of the Aortic Arch | |
|---|---|
|
Anomalies of the Thoracic Aorta
The nature of the embryologic development of the aortic arch and its branches leads to rather common malformations that can be clinically silent or can lead to clinical symptoms. Some of the anomalies are common, and others are quite rare (5,6). Table 19.2 outlines some of the common arch anomalies.
The left aortic arch with aberrant right subclavian artery is the most common major arterial anomaly, affecting 0.4% to 2% of the population. In this anomaly, the right subclavian artery takes off as the final branch of the aorta, not as the first branch. Patients with this anomaly are usually asymptomatic or have symptoms of dysphagia. There is no increase in the incidence of other associated congenital defects. Over half of these patients will show an abnormal mediastinal contour at the aortic arch level on frontal chest radiographs representing dilatation of the proximal portion of the aberrant artery, the so-called diverticulum of Kommerell (Fig. 19.4A). Lateral chest radiographs can show the abnormality as opacity projecting in the mediastinum behind the trachea, anterior to the spine, and above the aortic arch (the Raider triangle) (Fig. 19.4B) (7). The retrotracheal opacity represents the subclavian artery as it passes behind the esophagus and trachea through the mediastinum. If desired clinically, CT can prove the diagnosis of the aberrant artery (Fig. 19.4C). On occasion, the aberrant right subclavian artery can be seen as an oblique edge coming off the aorta and as an opacity projecting through the trachea extending to the right on the frontal view (Fig. 19.5) (8).
A right aortic arch identified incidentally in an adult almost always has an aberrant left subclavian artery.
Right aortic arch with mirror image branching has a very high (95%) association with severe congenital heart disease, including truncus arteriosus and tetralogy of Fallot.
The right aortic arch with aberrant left subclavian artery origin is an anomaly that is also generally an incidental finding on chest radiographs. However, there is a small association with other congenital heart defects. Chest radiographs show a right aortic arch
with opacity in the Raider triangle on the lateral view representing the aberrant artery (Fig. 19.6).
with opacity in the Raider triangle on the lateral view representing the aberrant artery (Fig. 19.6).
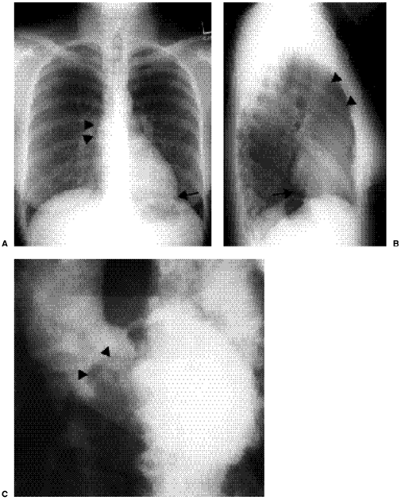 Figure 19.1 A 37-year-old patient with aortic stenosis and a bicuspid aortic valve. A. Posteroanterior chest radiograph shows a convex opacity along the right side of the mediastinum (arrowheads) representing poststenotic dilatation of the ascending aorta. In this case the left ventricle is enlarged, showing elongation of the heart and pointing of the cardiac apex toward the left costophrenic angle (arrow). B. Lateral chest radiograph demonstrates the dilated ascending aorta (arrowheads) and posterior displacement of the left ventricle at the posterior-inferior cardiac margin (arrow). C. Frame from a cineangiogram of the aortic root shows “doming” (arrowheads) of the stenotic bicuspid aortic valve. |
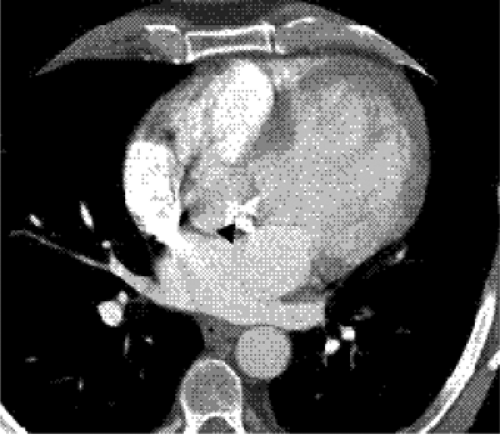 Figure 19.2 A 45-year-old woman with a bicuspid aortic valve and valve stenosis. Computed tomography image at the aortic valve level shows abundant calcification of the valve (arrowhead). |
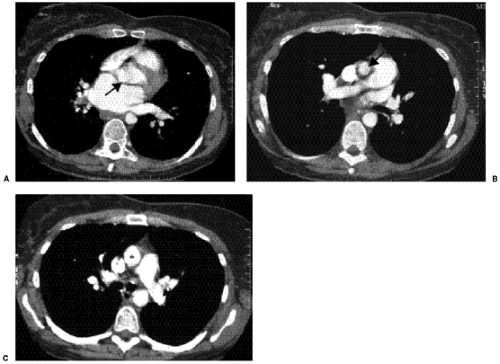 Figure 19.3 A 44-year-old patient with supravalvular aortic stenosis. A. Computed tomography image shows normal size and configuration of the aortic valve (arrow); (B) tubular narrowing of the supravalvular portion of the ascending aorta (arrow) and (C), normal diameter of the ascending aorta (asterisk). There is no poststenotic dilatation. |
The mirror image right aortic arch (no aberrant subclavian artery) has a high rate (95%) of association with severe congenital heart disease, usually of the cyanotic type such as tetralogy of Fallot, truncus arteriosus, or pulmonary atresia. However, the anomaly can be seen as an incidental finding on chest radiographs and CT studies. The double aortic arch forms a complete vascular ring, usually presenting in childhood with symptoms of compression of mediastinal structures. On occasion, the double arch first presents in adulthood (Fig. 19.7). The cervical aortic arch anomaly is relatively rare, usually presenting as an incidental finding (Fig. 19.8).
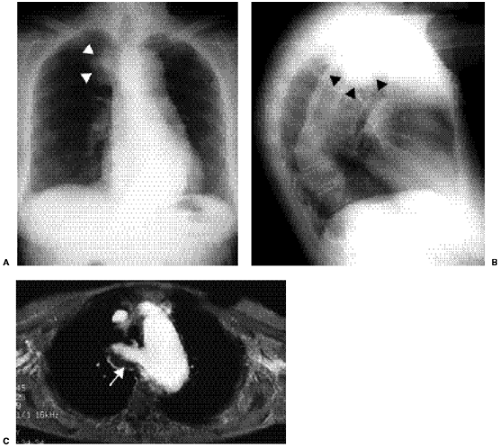 Figure 19.4 A 77-year-old patient with aneurysmal dilatation of the thoracic aorta and an aberrant right subclavian artery. A. Posterior anterior chest radiograph shows a left aortic arch. A mass-like protuberance projecting to the right of the superior mediastinum (arrowheads) represents an aneurysm of an aberrant right subclavian artery. B. The lateral chest radiograph shows an opacity posterior to the tracheal air column (arrowheads) in Raider’s triangle representing the aberrant subclavian artery shown on end. C. A computed tomography image of the arch demonstrates an aneurysm of the aberrant right subclavian artery. Thrombus is shown in the posterior portion of the aneurysm (arrow). |
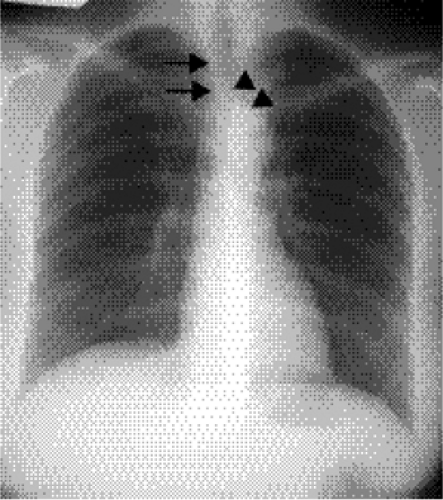 Figure 19.5 Posterior anterior chest radiograph shows an aberrant right subclavian artery as an oblique opacity (arrowheads) arising from the aortic arch, passing to the right. In this case the artery can be seen through the tracheal air column (arrows). |
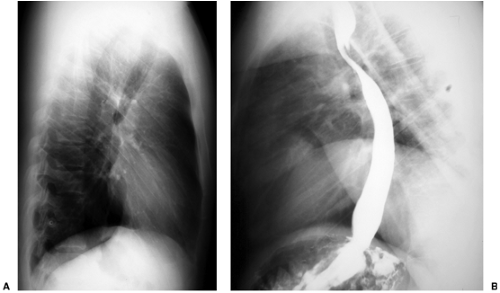 Figure 19.6 An adult patient with a right-sided aortic arch and an aberrant left subclavian artery. A. Lateral chest radiograph shows the aberrant left subclavian artery as a round opacity in Raider’s triangle. B. Lateral view of an esophagram shows a posterior impression on the barium column caused by the aberrant left subclavian artery. |
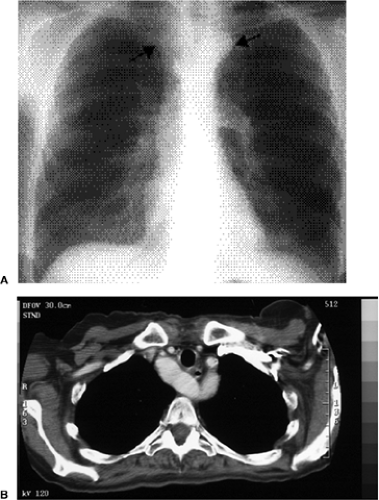 Figure 19.7 A 63-year-old patient with a double aortic arch. A. Posterior anterior chest radiograph shows a double aortic arch as opacities lying on both sides of the tracheal air column (arrows). B. Computed tomography image shows the double arches coming together posteriorly as a single descending thoracic aorta. The arches arose from a single ascending aortic lumen below this axial section. In this case, the arches are of equal caliber. The left arch is hypoplastic in many cases. |
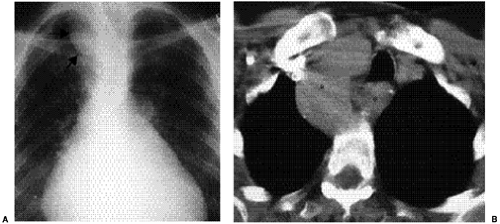 Figure 19.8 A 59-year-old patient with prior surgery for rheumatic heart disease and a right-sided cervical aortic arch. A. Posteroanterior view of the chest radiograph shows the high cervical right arch (arrows) at the thoracic inlet, displacing the trachea to the left. B. Computed tomography section shows the right cervical arch (a) at the thoracic inlet. There is an aberrant left subclavian artery (asterisk). |
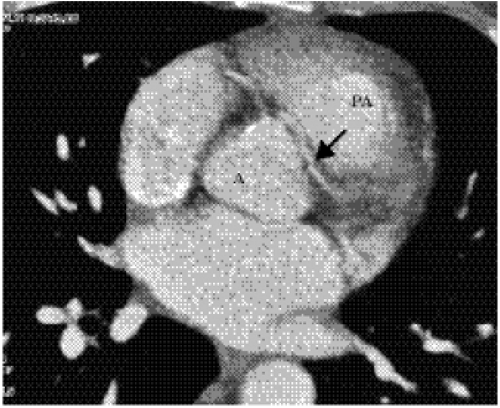 Figure 19.9 Computed tomography image shows an aberrant anterior descending coronary artery origin (arrow) arising from the right coronary sinus of Valsalva next to the right coronary artery. The left anterior descending artery passes between the aorta (A) and root of the pulmonary artery (PA). (Courtesy of James Mastromatteo, MD, Boston, MA.) |
Anomalous Coronary Artery Origins from the Aorta
Anomalous origin of a coronary artery can occur from the pulmonary artery or aorta. Patients with ectopic origin of both coronary arteries from the pulmonary artery are discovered early in life because of severe myocardial ischemia. Origin of one main coronary from the pulmonary artery with the other from the aorta is also a serious malformation, but some patients do survive past childhood. Ectopic origin of one or both coronary arteries, or of individual branches, from the aorta is compatible with life, found in 0.6% to 0.9% of the population (9,10). On occasion, CT or MRI can show these aberrant origins as an incidental finding. At other times, patients with unexplained chest pain and abnormal stress tests or a familial history of sudden death at a young age are referred for echocardiography, CT or MRI to show the coronary artery origins. The course of the ectopic coronary arteries can proceed from the origin to its myocardial distribution by several possible routes (11). The artery can pass between the pulmonary artery and aortic root (Fig. 19.9) or can course anterior or posterior to these structures. When a major coronary artery such as the left main or left anterior descending artery passes between the pulmonary artery and the aorta, there is a small risk of sudden death.
Anomalous coronary artery origin may be a cause of unexplained chest pain, particularly in young adults in whom atherosclerotic disease is less likely.
Left-to-Right Shunts
Atrial Septal Defect
The isolated small patent foramen ovale or atrial septal secundum defect is the most common postchildhood left-to-right shunt (12), with an estimated prevalence of 0.6 per 1000 (1). Ventricular septal defects are more common in children, but most close spontaneously or are repaired. Large atrial septal defects such as sinus venosus and ostium primum defects or atrial septal defects associated with other anomalies are usually discovered in childhood. Chest radiographs of patients with atrial septal defect show increased pulmonary vascularity and enlarged pulmonary arteries (Fig. 19.10). The heart can be normal in size but can enlarge, particularly in patients who develop mitral and tricuspid valve regurgitation (Fig. 19.11). Right ventricular enlargement is usually evident on the lateral view (Fig. 19.12). The aortic arch is relatively small in many patients with atrial septal defect. Cardiac MRI (Fig. 19.13) and CT (Fig. 19.14) can show the defect, although echocardiography is the primary modality used to confirm the diagnosis. Table 19.3 lists the common differential diagnosis for noncyanotic cardiac shunts.
An isolated arterial septal defect is the most common postchildhood left-to-right cardiac shunt.
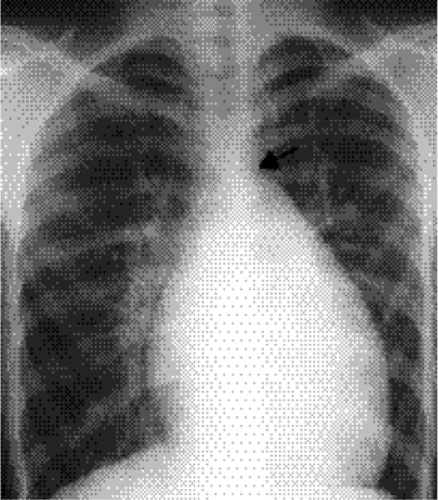 Figure 19.10 Posteroanterior chest radiograph shows increased pulmonary vascularity. The central pulmonary arteries are dilated. The rounded right heart border represents right atrial dilatation, and the round left heart margin is secondary to right ventricular enlargement. Many patients, like this one with atrial septal defect, have an associated small aortic arch (arrow). |
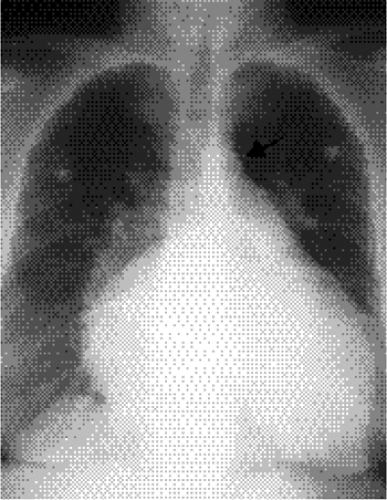 Figure 19.11 Posteroanterior chest radiograph shows markedly dilated chambers in this 76-year-old patient with an atrial septal defect. Echocardiography revealed tricuspid and mitral valve regurgitation. The aortic arch (arrow) shows atheromatous calcification but remains small in diameter. |
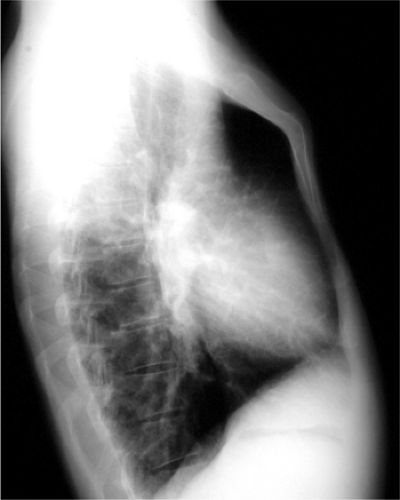 Figure 19.12 Lateral chest radiograph of a 49-year-old patient with an atrial septal defect shows fullness posterior to the sternum, representing right ventricular dilatation. There is pectus carinatum deformity of the sternum, an anomaly with known association with septal defects. |
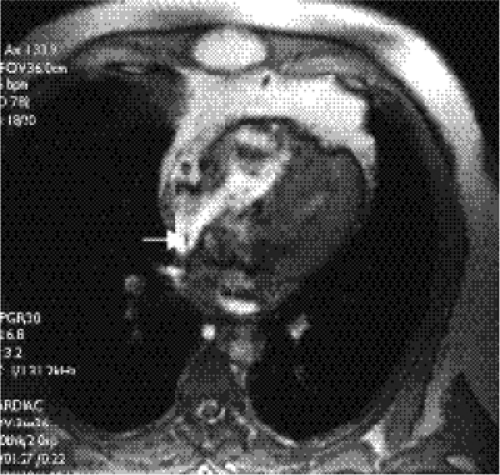 Figure 19.13 A 28-year-old patient with a patent foramen ovale. Frame from a cine-magnetic resonance imaging study in the four-chamber view shows a small dark jet of blood (arrow) crossing the interatrial septum from the left atrium (la) to the right atrium through the patent foramen ovale. |
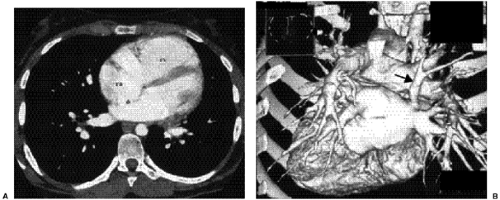 Figure 19.14 A 17-year-old girl with an atrial septal defect. A. Computed tomography section through all four cardiac chambers shows the atrial septal defect (asterisk) as interruption of the septum between the two atria. The right atrium (ra) and right ventricle (rv) are dilated, and the ventricular septum is flattened because of high pressure and volume in the right ventricle. B. Reconstructed three-dimensional image of the left atrium and pulmonary veins shows an anomalous course of a vertical vein emptying the posterior segment of the right upper lobe into the left atrium (arrow). Anomalies of pulmonary veins occur frequently in association with atrial septal defect, often emptying into the right atrium or vena cava. |
Ventricular Septal Defect
Isolated ventricular septal defects are relatively common congenital malformations in children. Many of these shunts close spontaneously. The estimated prevalence in adults is 0.3 per 1000 (1). Adults presenting with ventricular septal defects for the first time can be asymptomatic or can present with pulmonary hypertension and Eisenmenger physiology (13). Eisenmenger physiology occurs when there is pulmonary hypertension secondary to chronic increases in pulmonary blood flow and elevated right ventricular pressure.
Ventricular septal defects are uncommon in adults, usually either closing spontaneously (the majority) or having been closed surgically during childhood.
The elevated right ventricular pressure stifles left ventricular shunting and eventually leads to bidirectional flow across the septum, which in turn leads to cyanosis. Patients with small ventricular septal defects can have normal pulmonary vascularity and heart size. In these cases the diagnosis is made on physical examination and echocardiography. In patients with large shunts or Eisenmenger physiology, chest radiographs show increased pulmonary vascularity, enlargement of the central pulmonary arteries, right ventricular enlargement, and a normal aortic arch (Fig. 19.15). Left atrial enlargement is commonly seen in children but is usually absent in adults unless there is mitral valve regurgitation (14). MRI can show these defects in detail (Fig. 19.16).
Eisenmenger physiology with pulmonary hypertension develops secondary to chronically elevated pulmonary blood flow with elevated right ventricular pressure creating a bidirectional shunt and leading to cyanosis.
Table 19.3: Congenital Noncyanotic Cardiac Shunts | |
|---|---|
|
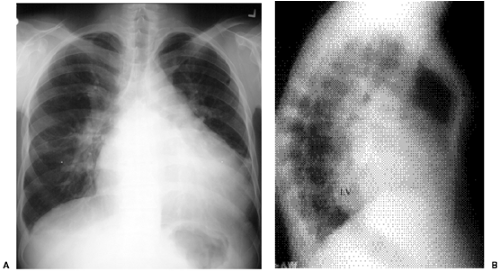 Figure 19.15 A 38-year-old patient with a 2:1 left-to-right shunt through a ventricular septal defect. A. A posteroanterior chest radiograph shows shunt vascularity, cardiomegaly, dilated central pulmonary arteries, and normal size of the aortic arch. B. A lateral chest radiograph on the same patient shows dilatation of the left ventricle (LV) posterior to the inferior vena cava. The dilated right ventricle is shown as fullness behind the sternum. This patient shows pectus carinatum deformity of the sternum (asterisk).
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|