Introduction and historic perspectives
“Is the inflammation of the heart always very sharp and acute, or does it not sometimes affect an insidious, hidden progress and which it appears, if not impossible, at least very difficult to distinguish?” Jean Corvisart, personal physician to Napoleon Bonaparte1
Myocarditis is usually defined by histologic criteria as inflammation of the myocardium, often with damage or injury to cardiac myocytes. The histologic Dallas criteria allow for two categories of myocarditis: active, in which myocyte damage is evident, and borderline, in which a cellular inflammatory infiltrate is not associated with myocyte injury or necrosis (Plate 48.1).2 However, the diagnosis of myocarditis is frequently presumed if a compatible clinical scenario is associated with new abnormalities on non-invasive cardiac imaging and/or elevated serum bio-markers of cardiac injury. Clinically, myocarditis most often presents as an acute or occasionally fulminant illness with new-onset dilated cardiomyopathy (DCM). However, manifestations range from subclinical disease to sudden death, new-onset atrial or ventricular arrhythmias, complete heart block or an acute myocardial infarction-type syndrome. The known causes of myocarditis are as varied as the disease presentations and include infectious causes (including coxsackie B virus, parvovirus B19, adenovirus, T. cruzi), toxic agents, and cardiac manifestation of a systemic autoimmune disease (Box 48.1).
DCM is a common phenotype that results from many forms of myocardial injury and is both an acute and long-term consequence of viral myocarditis.3 Usually a careful family history and a history of an antecedent viral prodrome in the weeks prior to cardiac symptoms permit the clinical distinction between familial and postviral cardiomyopathy. However, a small pedigree may confound the assessment of an inherited predisposition to DCM and inherited abnormalities of cytoskeletal proteins may actually predispose to more severe viral myocarditis, as is the case with Duchenne’s muscular dystrophy and coxsackie B virus infection, both of which affect dystrophin. Therefore a clear distinction between primarily familial and primarily acquired DCM is not always possible.
This chapter focuses on primary DCM in which histo-logic evidence of myocarditis or persistent viral infection is present. In the USA, the estimated prevalence of all forms of DCM is 36.5 cases per 100 000.4 DCM is important because it constitutes the leading cause (45%) of heart transplantation in the United States.5 In a review of 1230 cases of initially unexplained DCM, only 9% were thought to be due to myocarditis when the Dallas criteria were applied.6 Although standard histologic criteria for myocarditis are usually absent in chronic DCM, other markers of inflammation including ICAM, VCAM, and major histo-compatability complex (MHC) antigen expression are present in up to 40% of cases.7 Thus the prevalence of myocarditis in DCM varies widely depending on the histologic criteria used to establish the diagnosis.
Incidence, natural history and prognosis of acute myocarditis
The incidence of myocarditis in the community is unknown because many cases probably have minimal if any acute symptoms, and escape diagnosis only to present months or years later with a non-ischemic DCM. The incidence may also be underestimated due to the highly variable presentation of the disease which ranges from subclinical to fulminant heart failure, and includes sudden death, syncope, and acute myocardial infarction-like syndromes. Furthermore, the histologic diagnosis of patients with acute clinical symptoms and suspected myocarditis is confounded by a lack of sensitivity in the traditional Dallas Criteria.8 For example, in the Myocarditis Treatment Trial, which used the Dallas criteria for study entry, only 10% of 2333 patients with suspected myocarditis and recent-onset DCM met these criteria.9 The rate was somewhat higher at 17% in the European Study of Epidemiology and Treatment of Cardiac Inflammatory Diseases Study (ESETCID), which used less restrictive immunohistologic criteria.10
BOX 48.1 Select etiologies of myocarditis
Infectious
- Viral-common
- Parvovirus B19, increasing since 1994
- Adenovirus, most common 1995 to 2005
- Enterovirus (coxsackie B), decreasing since 2004
- HCV (in Japan)
- Human immunodeficiency virus
- Bacterial – consider in immunosuppressed hosts
- Mycobacteria
- Streptococcal sp.
- Trejxmema palladum
- Borrelia burgdoferi
- Fungal – consider in immunosuppressed hosts
- Aspergillus
- Candida
- Cryptococcus
- Histoplasma
- Coccidioides
- Protozoal
- Trypanosoma cruzi
Toxins
- Cocaine
- Anthracyclines
- Imatimib mesylate
- Interleukin-2
Hypersensitivity
- Clozapine
- Methyldopa
- Cephalosporins
- Penicillins
- Tricyclic antidepressants
Systemic/autoimmune
- Systemic lupus erythematosus
- Infammatory bowel disease
- Giant cell myocarditis
- Sarcoidosis (idiopathic granulomatous myocarditis)
- Takayasu’s arteritis
- Hypereosinophilic syndrome
- Endomyocardial eosinophilic diseases
The prognosis in myocarditis is variable and depends partly on clinical, hemodynamic, and histologic variables. Presentation with syncope, heart block on EKG, lower left ventricular ejection fraction, and higher pulmonary artery pressures are associated with shorter transplant-free survival.11 In the series from Massachusetts General Hospital, Dallas Criteria myocarditis was associated with shorter transplant-free survival than borderline myocarditis.12 In a Mayo Clinic series from the early 1990s by Grogan et al, the five-year survival in acute myocarditis was 56%, comparable to survival in patients with idiopathic DCM without myocarditis.13 Immunoperoxidase based diagnostic criteria have higher sensitivity than the Dallas criteria and may have prognostic value.14
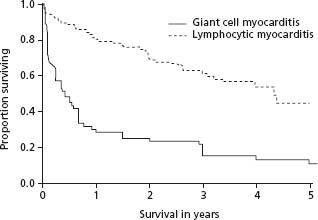
Histologic patterns that influence survival are limited to several uncommon disorders including giant cell myocarditis (GCM), acute necrotizing eosinophilic myocarditis, and cardiac sarcoidosis (CS).15 The five-year transplant-free survival rate in GCM is less than 20% (Fig. 48.1).16 In a large clinical series, subjects with either GCM or CS had a worse transplant-free survival than those with lymphocytic myocarditis.17 Paradoxically, patients with the clinical pathologic entity of fulminant lymphocytic myocarditis, although severely ill at presentation with marked hemodynamic compromise, are more likely to recover than those with acute (2 weeks to 3 months duration) myocarditis who do not have a distinct viral prodrome.18
Figure 48.1 Transplant free survival of lymphocytic myocarditis versus giant cell myocarditis. Kaplan–Meier curves showing the duration of transplant-free survival from the onset of symptoms in 63 patients with giant cell myocarditis and 111 patients with lymphocytic myocarditis from the Myocarditis Treatment Trial. P < 0.001 by log-rank test. (Reproduced with permission from Cooper et al.15)
A few serologic and imaging biomarkers have been associated with poor clinical outcome. Higher concentrations of serologic markers such as Fas ligand (FasL) and inter-leukin 10 (IL-10) may predict increased mortality.19,20 Right ventricular function on echocardiogram was an independent predictor of death or cardiac transplantation in a study of 23 patients with biopsy-confirmed myocarditis.21 In this study, multivariate analysis revealed that right ventricular dysfunction as quantitated by right ventricular descent was the most powerful predictor of death or cardiac transplantation. In a separate study, Naqvi et al demonstrated that contractile reserve assessed by dobutamine echocardiography predicted left ventricular functional recovery in 22 patients with new-onset DCM, some of which may have been due to myocarditis.22
Studies in mouse and rat models of myocarditis have investigated the pathogenesis of autoimmune and viral myocarditis and enhanced our understanding of the relationship between direct viral injury and the host immune response to viral infection. Coxsackie B virus-induced myocarditis (CVB) is thought to progress in three temporal stages:
1 acute myocarditis characterized by viremia and a high fatality rate in animal models
2 subacute myocarditis with lymphocytic infiltrates in the myocardium, rising antibody titers and release of inflammatory cytokines including IL-1-beta, tumor necrosis factor (TNF)-alpha, interferon (IFN)-gamma and IL-2
3 chronic myocarditis characterized by fibrosis, progressive ventricular dilation and heart failure.3
It is this third stage of “chronic myocarditis” that is thought to result in “idiopathic” dilated cardiomyopathy.
Myocardial injury in CVB and encephalomyocarditis virus (ECMV) myocarditis occurs through several mechanisms.23,24 Viral infection can lead within hours to days to myocyte death and release of sequestered intracellular antigens, which trigger an innate and adaptive immune response in the myocardium. Antibodies that cross-react with CVB, human cardiac myosin, and the beta-adrenergic receptor have been observed in cases of myocarditis. Cytokines, such as TNF-alpha, and autoantibodies associated with this inflammatory reaction impair cardiac myocyte contractility, and released cellular products such as major basic protein (MBP) can lead directly to myocyte cell death. T cell-deficient mice show a muted response or progression to myocarditis as compared to normal mice.25 Long-term myocardial damage may also occur through an apoptotic mechanism, especially in the absence of active inflammation. In the Lewis rat model of autoimmune myocarditis, T cells also are a key mediator of myocardial damage. Myocardial damage may occur independently of any immune reaction. For example, protein products of the enteroviral genome, including viral protease 2a, can cleave host proteins, including dystrophin, and lead to cardiomyopathy.26,27 Environmental variables such as selenium deficiency can affect viral virulence and individual susceptibility to cardiotopic viral infection.28,29
Genetic susceptibility to myocarditis is strongly suggested by animal models in which spontaneous myocarditis occurs.30 A genetic component of DCM has also been well characterized in studies of familial association and linkage studies. Twenty to 40% of patients with DCM have a first-degree relative with some degree of left ventricular dysfunction.31 A positive association of DCM with the MHC HLA DR4 has been identified in several independent studies. However, specific genes that predispose patients to myocarditis have not yet been identified.
Clinical presentation and diagnosis
The clinical presentation in myocarditis varies from asymptomatic ECG findings to cardiogenic shock or even sudden death. Although a non-specific viral prodrome with fevers, myalgias, and respiratory or gastrointestinal symptoms is classically associated with myocarditis, the published rates of reported symptoms are highly variable.32 Cardiac symptoms include fatigue, decreased exercise tolerance, palpitations, precordial chest pain, and syncope. Of the 3055 patients in the ESETCID study, 72% had dyspnea, 33% had chest pain, and 18% had arrhythmic events.10 Most studies of acute dilated cardiomyopathy and suspected myocarditis have a slight a male predominance. For example, 62% of the 111 patients enrolled in the Myocarditis Treatment Trial were male.9
Cardiac enzyme elevations are seen in a minority of patients with acute myocarditis and can help confirm the diagnosis. Standard markers of myocardial damage including troponin-I have a high specificity (89%) but limited sensitivity (34%) in the diagnosis of myocarditis, especially with a shorter duration (less than four weeks) of symptoms.33 Clinical and experimental data suggest that cardiac troponin-I is increased much more frequently than creat-nine kinase MB (CK-MB) in patients with myocarditis.34 Other circulating biomarkers including cytokines, complement components and anti-heart antibodies have not been prospectively validated and are not in widespread clinical use.
Troponin I or T should be measured in suspected acute myocarditis (Class I, Level C).
The ECG in acute myocarditis may show sinus tachycardia with non-specific ST segment and T-wave abnormalities. Occasionally, the ECG changes are suggestive of an acute myocardial infarction and may include ST segment elevation in two or more contiguous leads (54%), widespread ST segment depressions (18%) and pathologic Q-waves (18–27%).35 In a small proportion of patients, various degrees of heart block may occur. In the Myocarditis Treatment Trial, pacemaker implantation occurred in approximately 1% of subjects. Ventricular arrhythmias may also be present, but occur more commonly in cardiac sar-coidosis and giant cell myocarditis.36
An ECG should be performed in suspected acute myocarditis (Class I, Level C).
The most common echocardiographic features of acute myocarditis are unfortunately not specific; however, echocardiography is useful to exclude other causes of acute heart failure. Echocardiographic patterns of dilated, hypertrophic, restrictive, and ischemic cardiomyopathies have been described in histologically proven myocarditis. Segmental or global wall motion abnormalities can simulate myocardial infarction in myocarditis.37 In the Myocarditis Treatment Trial, increased sphericity and left ventricular volume occurred in acute, active myocarditis. Left ventricular cavity size may be normal in very early myocarditis and increase over time due to remodeling. Fulminant myocarditis may be distinguished from acute myocarditis by echocardiographic criteria as suggested by Felker et al.38 These authors performed echocardiography on 11 fulminant and 43 acute myocarditis patients at presentation and after six months. Patients with fulminant myocarditis had near normal LV diastolic dimensions with increased septal thickness at presentation, while those with acute myocarditis had increased diastolic dimensions but normal septal thickness.
An echocardiogram should be performed in suspected acute myocarditis (Class I, Level C).
Due to its availability and low risk, cardiac magnetic resonance imaging (CMR) is being used with increasing frequency as a diagnostic test in suspected acute myocarditis and may be used to localize sites for endomyocardial biopsies.39,40 CMR may also be useful to differentiate ischemic from non-ischemic cardiomyopathy. McCrohon et al performed gadolinium-enhanced CMR in 90 patients with heart failure and LV systolic dysfunction, of whom 63 had idiopathic DCM. Subendocardial or transmural enhancement was seen in ischemic cardiomyopathy. In contrast, the DCM group had three patterns of enhancement: no enhancement, myocardial enhancement indistinguishable from the patients with CAD and patchy or longitudinal mid wall enhancement.41 Results from similar studies suggest that diffuse and/or heterogeneous involvement with combined enhancement is highly suggestive of myocarditis. Histopathologic evaluation of biopsies directed by contrast CMR with delayed enhancement by Mahrholdt et al demonstrated active myocarditis in 19 of 21 patients. In contrast, only one in seven patients showed active myocarditis in biopsies guided by non-myocardial delayed enhancement (MDE) CMR.42 Serial CMR using T1-weighted images with gadolinium have also been used to visualize the myocardial injury in myocarditis and track its progression.43 The prognostic value of CMR to predict future cardiac events in acute myocarditis is under investigation.
CMR can be useful in suspected acute myocarditis (Class IIa, Level C).
The gold standard for the diagnosis of myocarditis is histopathology on endomyocardial biopsy (EMB). The histopathologic classification called the Dallas criteria44 was used in the Myocarditis Treatment Trial and is widely cited, but the average incidence of positive right ventricular biopsy findings in acute dilated cardiomyopathy is only around 10%.9,45 This is likely due to sampling error secondary to the patchy nature of myocardial inflammation46 and interobserver variability.8,47 Newer diagnostic criteria that rely on immunoperoxidase techniques are coming into broader clinical use. Expression of MHC antigens is a more sensitive marker than the Dallas criteria for myocardial inflammation, and this is detected using immunoperoxi-dase-based stains for HLA-ABC and HLA-DR antigens. Indeed, the sensitivity and specificity of MHC expression for detecting biopsy-proven myocarditis has been reported as high as 80%. Viral genomes can be detected using molecular methods like the polymerase chain reaction (PCR) to identify viral RNA and DNA from endomyocardial biopsy samples. The clinical value of viral genome analysis, immunoperoxidase stains, and transcriptome based biomarkers is being evaluated in ongoing clinical trials.
Currently, the risks of cardiac perforation and death associated with EMB preclude its routine use in the diagnosis of myocarditis. The 2007 AHA/ACCF/ESC scientific statement on the role of endomyocardial biopsy in cardiovascular disease lists 14 clinical scenarios in which EMB may be considered, and gives a class I recommendation to two of these 14 (Table 48.1).49
Table 48.1 The role of endomyocardial biopsy in 14 clinical scenarios
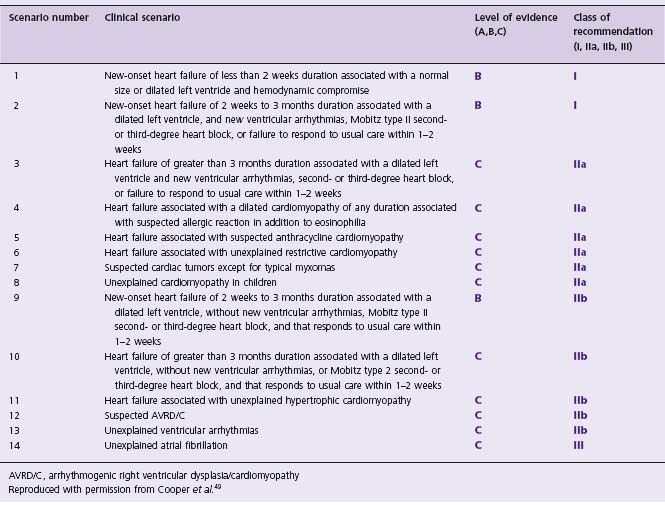
AVRD/C, arrhythmogenic right ventricular dysplasia/cardiomyopathy
Reproduced with permission from Cooper et al.49
Myocarditis presenting as acute DCM
Myocarditis should be treated according to the guidelines for the presenting clinical syndrome. For example, acute DCM should be managed according to the current AHA/ ACCF, ESC, and HFSA guidelines.48–51 The mainstay of therapy for acute myocarditis is supportive therapy for left ventricular dysfunction. Most patients will improve with standard heart failure regimen that includes ACE inhibitors or angiotensin receptor blockers, beta-blockers such as metoprolol or carvedilol, and diuretics, if needed. In patients who deteriorate despite optimal medical management, case series suggest a role for mechanical circulatory support such as ventricular assist devices or extracorporeal membrane oxygenation (ECMO) as a bridge to transplantation or recovery.52,53
Heart failure therapy
Angiotensin-converting enzyme inhibitors/angiotensin receptor blockers (ACEI/ARBs)
Although the utility of ACEI/ARBs in the treatment of symptomatic and asymptomatic heart failure with left ventricular dysfunction is well established, evidence for their specific use in myocarditis is limited to studies in animal models. Early treatment with captopril, starting on day 1 of infection, has beneficial effects in murine models of myocarditis in the reduction of inflammatory infiltrates, myocardial necrosis and calcification.54 In a second study by Rezkalla et al, delayed captopril therapy, initiated 10 days after viral inoculation, was still found to result in a reduction of left ventricular mass and liver congestion.55
Studies of the ARB candersartan in a murine model of viral myocarditis showed a significant improvement in seven-day survival (60% versus 18%) in the candersartan group as compared to controls.56
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


