Chapter 46 Ventricular Fibrillation
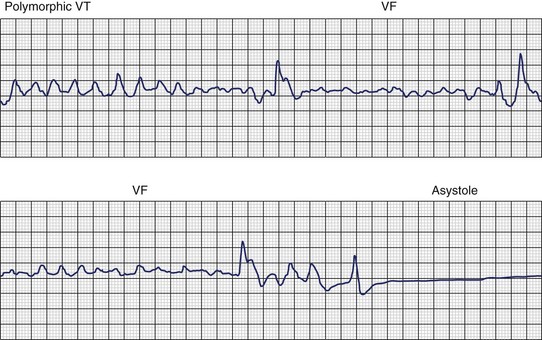
Ventricular fibrillation (VF) is the most serious cardiac arrhythmia and has a primary role in mediating sudden cardiac death (SCD). It leads to immediate circulatory arrest with cardiovascular collapse. A variable period may elapse, but cardiac asystole usually supervenes (Figure 46-1). Spontaneous termination of VF, which is seen in animal experiments, is rare in humans. VF is often preceded by an organized, rapid ventricular tachycardia (VT) of variable duration; recordings from implantable devices have now substantiated this. Ischemic injury may trigger VF. If untreated, this leads to irreversible end-organ damage, including cerebral and myocardial damage after 5 to 7 minutes of VF. Even with optimally performed basic cardiopulmonary resuscitation (CPR), the mortality rate is greater than 95% if defibrillation is delayed by more than 10 minutes. The rule of survival from out-of-hospital VF is that survival decreases by 10% for each minute before defibrillation. The sine qua non of effective resuscitation from VF is thus prompt defibrillation, delivered as early as possible. Importantly, however, a brief period of CPR before defibrillation in out-of-hospital cardiac arrest, especially if the arrest is more than 4 minutes in duration, may increase survival rates.
Etiology
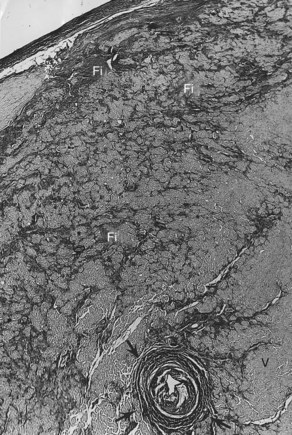
VF occurs in many disease states of the myocardium and the conduction system and can be broadly grouped under two categories: (1) genetic/familial and (2) acquired. Genetically based abnormalities of the myocardium or the specialized conduction fibers or both may give rise to the clinical manifestations of familial occurrence of VT and VF. In most cases, the initial arrhythmia is one of various forms of VT (monomorphic or polymorphic VT, which then degenerates to VF). However, acquired cardiac diseases such as coronary artery disease (CAD), hypertensive heart disease, cardiomyopathy of any etiology with or without heart failure, and other miscellaneous disease states (see Chapter 47) are the most common causes of VF and SCD.1–3 VT leading to VF may occur in acute myocarditis of any etiology. However, intractable VT with recurrent cardiac arrest refractory to medications or defibrillator device implantation may be related to chronic myocarditis (Figure 46-2).4–6
Pathology
The anatomic basis for VF in acquired heart diseases is similar to that of VT. In this chapter, emphasis is given to the morphologic findings for VF in athletes or healthy youths who were seemingly living a “normal, asymptomatic” life and died suddenly. In addition, familial occurrences of SCD caused by recurrent VT degenerating to VF is briefly discussed.1–3,5,6
Sudden Cardiac Death in Athletes or the Young and Healthy
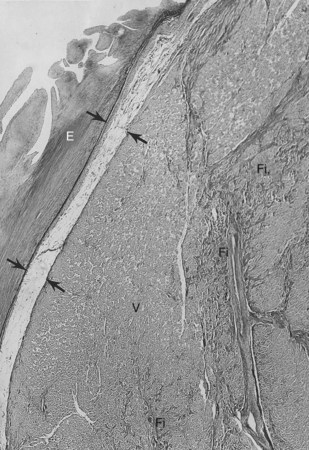
The conduction system has been carefully studied in several young persons who were living a “normal” life and died suddenly. Routine autopsies are often unremarkable. In the majority, the heart is hypertrophied and enlarged to a mild, moderate, or marked extent; however, at the gross level, no significant abnormalities were seen.3 A serial section examination of the conduction system and the surrounding myocardium reveals varying types of abnormalities, either of a congenital or acquired nature. Congenital abnormalities can exist in the sinoatrial (SA) node, atrioventricular (AV) node, or the AV bundle and the bundle branches (Figure 46-3). Abnormal formation of the SA or AV nodes, such as a double SA node, a double AV node, or the abnormal location of the SA and AV nodes in unexpected areas, can be seen pathologically. The AV bundle may be considerably fragmented into several components, abnormally located, or both. In addition, acquired pathologic findings exist in the form of focal myocardial disarray, fat, or fibrosis to varying degrees, disrupting or replacing parts of the specialized conduction fibers and the surrounding myocardium (see Figure 46-3) with arteriolosclerosis (small vessel disease) of the ventricular septum. In general, the findings in the conduction system are accompanied by pathologic findings in the surrounding myocardium. Mononuclear cell infiltration in the approaches to the SA node and in the SA node itself is present.3
Despite such pathologic findings in and around the conduction system, these persons were considered to be asymptomatic, and SCD was the first manifestation of the disorder. Clinically, in almost all, VF was the only common denominator observed by the paramedics at the time of resuscitation. It can be hypothesized that varying types of congenital or acquired pathologic findings in and around the conduction system may remain “silent,” and these individuals are asymptomatic for long periods. Other disorders (e.g., right ventricular cardiomyopathy, hypertrophic cardiomyopathy, idiopathic dilated cardiomyopathy) can lead to VT degenerating to VF, the first evident manifestation of the disease. During an altered physiological or metabolic state, anatomic findings, pathologic findings, or both may trigger an arrhythmia that progresses to VT, VF, and SCD.3
Familial Sudden Cardiac Death
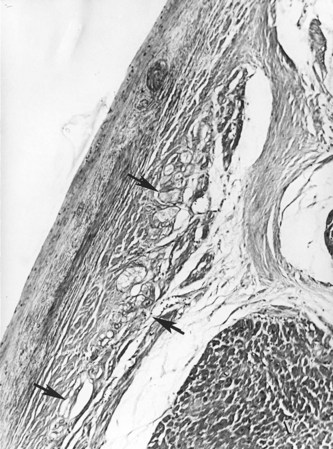
A genetic tendency for the development of an abnormal conduction system, the surrounding myocardium, or both may also lead to VT, VF, and SCD (Figure 46-4).5,6 Familial occurrence of ventricular arrhythmias may be related to the many genetic mutations associated with congenital long QT syndrome (LQTS) or Brugada syndrome. These disorders of ion channel function may also lead to cellular dysfunction and pathologic changes such as fatty infiltration, fibrosis, and disruption of the conduction system and the adjoining myocardium.7,8 These disorders are discussed in other chapters in this text and are alluded to in subsequent sections of this chapter.
Epidemiology
Ventricular Fibrillation and Sudden Cardiac Death
The epidemiology of VF intertwines with the available data on SCD (or cardiac arrest) and its documentation by emergency medical system (EMS) personnel.9 In 1998, an epidemiologic prospective study from northeast Italy reported an incidence of cardiac arrest of 95 of 100,000 persons per year.10 In this study, VF accounted for 30.2% of the initially recorded rhythms, asystole for 48.3%, and pulseless electrical activity for 21.5%. A similar study of out-of-hospital cardiac arrest confirmed VT or VF as the initial rhythm in 59 (30%) of 197 patients of a patient cohort from the Los Angeles area.11 Although VT and VF were consistently more likely to be associated with return of spontaneous circulation in these two studies, the time dependency of the initial recorded rhythm is still debated. The Ontario Prehospital Advanced Life Support (OPALS) study quoted an annual incidence of 58 out-of-hospital cardiac arrests per 100,000 persons, and in the subgroup of EMS-witnessed cardiac arrests, VT and VF accounted for 34.2% of cases.12 However, a recent analysis from Sweden estimated that patients with an electrocardiogram (ECG) taken within the first 10 minutes from a witnessed cardiac arrest have an incidence of VF of 50% to 60%. Linear regression further estimated that ECGs taken within the first 4 minutes should have an incidence of 75% to 80%.13
Ventricular Fibrillation and Population Considerations
Despite the debate on the true proportion and incidence of VF in prehospital cardiac arrest, event rates range from 250,000 to more than 450,000 per year in the United States.14 This absolute number is heavily influenced by population dynamics and the defined subpopulation being discussed.
The median incidence of cardiac arrest in 10 large Canadian and U.S. communities is 52 cases per 10,000 annually.15 The well-documented high-risk subgroups (ischemic heart disease with low left ventricular ejection fraction [LVEF], complex ventricular ectopy, prior hospital admission for congestive heart failure [CHF], or previous cardiac arrest) contribute a minority of the total number of cardiac arrests, although the subgroup percent incidence per year is the highest (Figure 46-5).
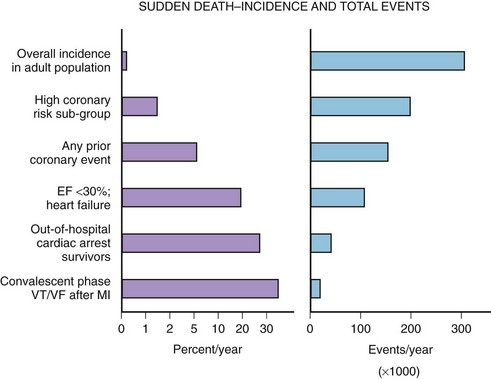
A screening tool or preventative intervention would need to be applied to 999 of 1000 persons to influence the 1 of 1000 previously unidentified persons destined for a cardiac arrest. The Framingham study demonstrated a large disparity from the highest to lowest decile (14-fold increase in risk) for SCD measuring the well-known risk factors such as age, family history, gender, tobacco use, hyperlipidemia, and hypertension.16 Electron beam computed tomography for the detection of coronary artery calcification has been correlated to coronary event risk.17 A recent publication compared the Framingham risk index and coronary artery calcification in an autopsy series of SCD victims.18 The two risk assessment methods had modest correlation with each other (63%). Of the cases, 83.5% had a coronary artery calcification score or Framingham risk index above average for age. The remaining 17.5% highlight the need for exploration of new cardiac risk factors (e.g., fibrinogen, homocysteine, infectious agents, or ion channel abnormalities) that could be applied to the population at large.
The high-risk subpopulations have been targeted in recent publications for epidemiologic analysis. The Worcester Heart Attack Study described a relatively fixed incidence rate of 4.7% for VF in hospitalized patients with validated acute myocardial infarction (MI) over a 22-year period (1975 to 1997).19 The Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico (GISSI-2) database reported an incidence of early-onset VF (<4 hours after onset of acute MI) and late VF (>4 to 48 hours) of 3.1% and 0.6%, respectively. In-hospital prognosis was worse in patients with VF than without VF, but the postdischarge to 6-month mortality rate was similar for both VF subgroups and controls.20 The patient with prior cardiac arrest has a similar “time dependence” of risk of recurrent events, as was presented by Furukawa et al (Figure 46-6).21
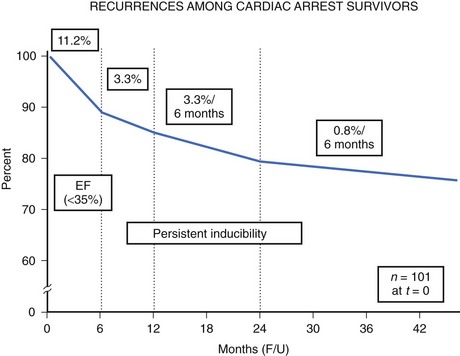
Low LVEF was a strong predictor of the highest risk of recurrence (11% in the first 6 months). Persistent inducibility at electrophysiological study (EPS) was the better predictor for recurrent events from 6 to 42 months, but the subsequent risk fell to 0.8% over the final 6 months of the study period. In the patients who entered the Antiarrhythmics Versus Implantable Defibrillators (AVID) study with VF, the survival curve shows the same early recurrence (<6 months) in the patients treated with antiarrhythmic drugs as those who did not receive this therapy (Figure 46-7).22
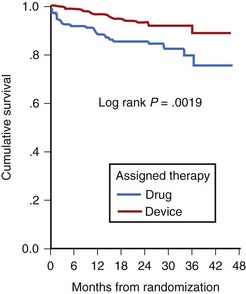
Clinical trials are assessing the “static” risk of cardiac events outside the setting of acute hospitalization.23 The Multicenter Automatic Defibrillator Implantation Trial (MADIT-II) trial tested implantable cardioverter-defibrillator (ICD) therapy versus standard medical therapy in patients with ischemic heart disease, an LVEF less than 30%, and previous MI. It showed a 31% relative risk reduction with ICD therapy. The Sudden Cardiac Death in Heart Failure Trial (SCD-HeFT) trial compared three arms of therapy in patients with congestive heart failure and LVEF less than 30%: best conventional heart failure therapy with placebo, best conventional heart failure therapy with amiodarone, and best conventional heart failure therapy with ICD.23 In a preliminary report, there was a 23% relative reduction in all-cause mortality with the ICD compared with placebo (7.2% to 5.8% per year), and no reduction in mortality rate with amiodarone compared with placebo. The most recent developments in assessing risk of cardiac arrest have been in the blossoming technology of molecular genetics. The familial types of LQTS and their respective genetic bases have been well described elsewhere. An increasingly recognized syndrome of right bundle branch block (RBBB), ST segment elevation, and aborted SCD (Brugada syndrome) has also been ascribed to a genetic mutation in the gene encoding the cardiac sodium channel. Its prevalence in a European study by ECG screening was estimated at 0.1% of the general population.24 If the only effective therapy for the syndrome is ICD implantation, the economic impact on the public health budget is obvious.
A trial of the automated external defibrillator (AED) to be used in the home by trained spouses or cohabitants of individuals with prior anterior MI (the Home Automated External Defibrillator Trial [HAT]) was not able to show a reduction in mortality rate, in part because of the unexpectedly low incidence of VF as the initial recorded arrhythmia in these patients.25
Basic Electrophysiology
Nature of Fibrillatory Wavefronts
Re-entrant excitation is the fundamental mechanism responsible for VF. The precise nature of these wavefronts, their formation, and sustainability are key to understanding VF. Various theories exist regarding the nature of re-entrant waves. One such theory is the leading circle hypothesis, originally put forward by Allessie and coworkers to explain atrial fibrillation (AF).26 This hypothesis states that a re-entrant circuit is the smallest pathway in which the impulse can continue to circulate and that the core is kept permanently refractory (i.e., there is no excitable gap) (Figure 46-8). However, Janse questioned whether the core is truly refractory during re-entry, since membrane potential is relatively normal in this region and diastolic intervals are relatively long.27 Furthermore, the presence of an excitable gap allows wavefronts (spontaneous or induced) to invade the area and terminate or entrain the re-entrant circuit; Allessie et al were able to terminate tachycardia with premature beats (i.e., anti-tachycardia pacing).26 It now appears that in most cases of VF, an excitable gap does exist; however, there may still be room for leading circle re-entry in a subset of arrhythmic phenomena, particularly with regard to AF.28
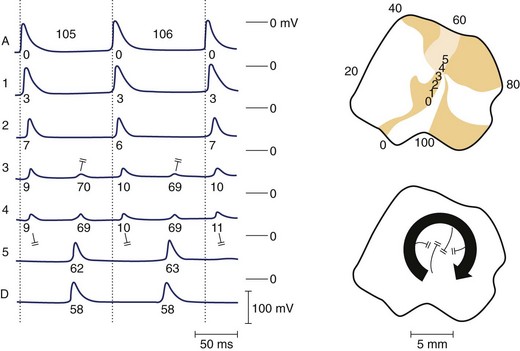
An alternative mechanism for VF is spiral wave re-entry. This theory suggests that the wavefront curves, or forms a spiral, because curvature is negatively related to conduction velocity (because of the source-sink relationship). As a result, the wave is highly curved near the core and moves slowly, but at the distal end the wave speed increases, resulting in a spiral shape. Two-dimensional spiral waves may theoretically exist in surviving two-dimensional layers overlying the healed myocardial infarcts, although three-dimensional waves (i.e., scroll waves) are clearly more relevant to cardiac arrhythmias. A scroll wave can be thought of as a stack of spiral waves in which the cores line up to form a “filament” at the center. Simple scroll waves, in which the filament forms a straight line, have been demonstrated during VF.29 A scroll wave with a core that is perpendicular to the recording surface will appear as a two-dimensional spiral wave, whereas a filament that is parallel to the recording surface will appear as a plane wave. It has been suggested that during VF, scroll waves are usually oriented parallel to the surface, which may explain why mapping studies sometimes fail to detect re-entry during VT or VF. A scroll wave with a ring-shaped filament is called a scroll ring. It is interesting to note that the cross-section of such a wave would represent classic figure-of-eight re-entry. Only indirect experimental evidence is available for the existence of scroll rings, probably because of the difficulty of measuring them, because they manifest only transmurally, never on the surface.30 The shape of a scroll as it is initiated strictly depends on the shape of the inexcitable obstacle encountered by the wavefront. Thus, filaments of varying shape can readily develop in the myocardium. Scroll rings ultimately collapse on themselves, but simple scroll waves can be stabilized and maintained.31
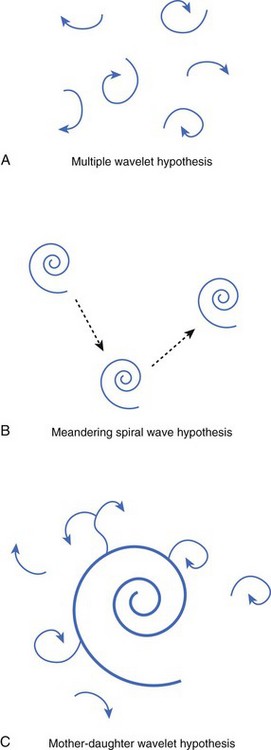
Numerous hypotheses have been offered regarding the nature of fibrillatory waves that underlie VF. Three of these theories are illustrated in Figure 46-9: (1) the multiple wavelet hypothesis, (2) the meandering spiral wave hypothesis, and (3) the mother-daughter wavelet hypothesis. The multiple wavelet hypothesis was put forward in the early 1960s by Moe and describes fibrillation as consisting of multiple, nonstationary wavefronts that continuously form, fractionate, and reform (Figure 46-9, A).32 Spatial dispersion of repolarization was an important condition for initiating multiple wavelet VF. Others have suggested that VF is caused by spiral re-entrant waves that meander around the myocardium (see Figure 46-9, B).33 According to this theory, the chaotic appearance of the ECG in VF is attributable to the meandering of a single spiral wave rotor throughout the ventricular myocardium. Jalife suggested that the mechanism of fibrillation may consist of a stable, periodic re-entrant wave that gives off “daughter wavelets” that meander and fractionate (see Figure 46-9, C).34 According to this hypothesis, unstable daughter wavelets form because of local gradients of refractoriness and give rise to the fibrillatory patterns of the ECG.
Substrates for Ventricular Fibrillation: Heterogeneities of Repolarization and Refractoriness
Various anatomic and functional substrates can lead to the development of the fibrillatory dynamics previously described. Early studies of AF suggested that the heterogeneity of refractoriness was a necessary substrate for fibrillation in the heart.32 Although it was later shown that VF can occur in hearts without large dispersions of refractoriness (i.e., in normal hearts), or in modeling studies using simulations of electrically homogenous tissue, these studies may not be clinically relevant.35 It is more difficult to initiate VF in a normal heart, and the vast majority of fibrillatory episodes occur in diseased hearts, in which the gradients of repolarization kinetics (and therefore refractoriness) are almost certainly abnormal. The important role of repolarization in the development of re-entrant substrates has recently been emphasized by studies demonstrating the inherent heterogeneities in repolarization properties that exist transmurally.36 Antzelevitch and others have demonstrated that trans-mural heterogeneities of repolarization are critical in the development of the substrates for re-entry under various drug-induced or disease-induced conditions.37,38
Rosenbaum et al recently developed a hypothesis that explains how the critical heterogeneities of repolarization that provide a substrate for re-entry may occur.39 Beat-by-beat alternation of cardiac action potential duration (APD) has been shown to underlie T-wave alternans, an electrocardiographic indicator that correlates well with the risk of SCD.39 We have demonstrated that when alternans occurs in various regions of the myocardium in a “discordant” manner (i.e., some regions are on a long-short-long cycle, and others are on a short-long-short cycle), steep gradients of repolarization develop and reverse the direction on a beat-by-beat basis, which can lead to conduction block, re-entry, and VF.40
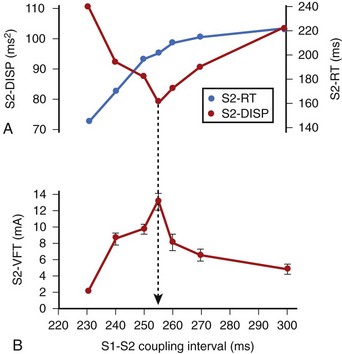
In related work, Laurita et al demonstrated that premature beats modulate the dispersion of repolarization in a manner that has direct effects on vulnerability to VF.41 As the S1-S2 interval is shortened, gradients of repolarization decrease concomitant to a decrease in vulnerability to VF, but then increase with a concomitant increase in VF vulnerability as S1-S2 is further shortened (Figure 46-10). Such biphasic modulation of dispersion relates back to the concept of the “vulnerable period,” which arose from critical studies by Moe, who described the window of time during the ECG T wave when vulnerability to VF is greatest.32 Thus, dispersion of repolarization and refractoriness appears to play a major role in the mechanism of re-entrant VF.
Restitution Hypothesis
Restitution is a property of cardiac myocytes that dictates the APDs of a premature action potential after a period of steady-state rhythm. The APD of the extrasystolic beat is determined not only by the S1-S2 interval but also by the APD of the pre-extrasystolic beat. Furthermore, gradients of restitution kinetics are known to exist across the epicardial surface such that cells in different regions of the heart having equal baseline APDs could have different extrasystolic APDs following the same S1-S2 interval.42 In theory, if gradients of restitution kinetics are steep enough, underlying heterogeneities of repolarization would not be necessary to create dispersions of repolarization following a premature beat.42 However, cardiac tissue is known to exhibit repolarization heterogeneities across epicardial and trans-mural surfaces, so the role of restitution may be to enhance existing dispersion of repolarization and promote VF.
One possible mechanism to describe the role of restitution in VF is the restitution hypothesis, which states that when the restitution curve (a plot of APD vs. diastolic interval) has a slope greater than 1, VF may be initiated. For example, a wavefront encroaching on the tail end of a previous wave creates a gradient of diastolic interval between the two (assuming the degree of curvature is not identical); in regions of tissue where restitution is steep, this will create large dispersions of APD along the second wavefront. Some areas of the wave may have such a short diastolic interval that they fail to propagate, resulting in wavefront fractionation. This was demonstrated by Garfinkel et al, who also used pharmacologic agents to flatten the restitution curve slope and decrease vulnerability to VF. Koller et al showed that elevating extracellular potassium decreases the portion of the restitution curve where the slope is 1 or more and converts VF to a periodic rhythm.43,44 Other investigators have shown that the biphasic nature of a restitution curve, demonstrated in a number of cardiac tissue types, is also an important determinant of vulnerability to VF.45 More recently, the restitution hypothesis has been applied to restitution curves for both APD and conduction velocity.46
Myocardial Ischemia
VF is commonly caused by acute myocardial ischemia. Under experimental conditions, it is often difficult to provoke VF in normal tissue using premature stimuli; however, in ischemic tissue, premature beats exacerbate dispersion of refractoriness and readily lead to tachyarrhythmias.47 This is because of the various electrical alterations that occur during ischemia and the consequent formation of various substrates that promote re-entry and VF.
At the cellular level, acute ischemia results in depolarization of the resting membrane potential, decreased maximum rise rate and amplitude of the action potential, and decreased APD, as well as reduced intercellular coupling.48–53 Depolarization of the membrane is attributed largely to accumulation of extracellular potassium, as is post-repolarization refractoriness.54,55 Acidosis can produce a small depolarization of resting membrane potential and also decreases gap junction conductance and cell-to-cell coupling.56 The net result of the electrical changes that occur during ischemia is the formation of various substrates that can lead to re-entry and wavebreak, such as slowed conduction velocity, increased dispersion of refractoriness, and alternans.
Healed Myocardial Infarcts
The process of tissue healing and scar formation after an episode of acute myocardial ischemia involves necrosis of the infarcted region as well as swelling and hypertrophy of the noninfarcted region as it attempts to compensate for the loss of cardiac muscle.57 Heterogeneity of repolarization may result from the uneven prolongation of APD in the hypertrophied post-MI ventricle, changes in expression of gap junction proteins, or both.58,59 A healed infarct may also provide an anatomic substrate for arrhythmias. Infarcts vary from simple surviving muscle bundles that form accessory pathways to complex subendocardial sheetlike structures, which are linked to the surrounding myocardium by multiple connecting bundles, creating complex matrices of conductive tissue that may promote multiple re-entrant circuits. In particular, the combined effects of slowed conduction and the presence of structural anomalies is significant because in tissue where excitability is low, a wavefront breaking past an obstacle will curl at the inner ends (where the break occurred) to create figure-of-eight re-entry, whereas in a normally excitable medium, the wave tends to reform on the other side of the obstacle.60
Autonomic Modulation of Ventricular Fibrillation
It has been well established that increases in sympathetic tone increase the risk for SCD, whereas vagal reflexes have the opposite effect.61–63 Studies in dogs have shown that left cardiac sympathetic denervation, or left stellectomy, increases survival and decreases arrhythmias during acute MI.64 In contrast, unilateral blockade of the right stellate ganglion by cooling increases the number of arrhythmias after coronary occlusion.65 Many clinical trials have shown that β-adrenergic receptor blocking drugs have a potential benefit in preventing SCD in patients with MI, whereas anti–α-adrenergic agents have not been shown to be effective.66–69
It has been suggested that during acute MI, sympathetic nerves that traverse the myocardial wall are damaged, leading to denervation hypersensitivity.70 This, in turn, leads to spatially inhomogeneous responses to β-adrenergic stimulation across the ventricle, forming potential substrates for re-entrant excitation. Further investigation is required to elucidate fully the mechanistic role of β-adrenergic stimulation and arrhythmogenesis.
Genetic Substrates for Ventricular Fibrillation
Brugada syndrome is a cardiac disease characterized by an elevated ST segment that is unrelated to ischemia, Q-T interval prolongation, electrolyte abnormalities, or structural heart disease.71 Patients with Brugada syndrome are at increased risk of SCD from VF. The syndrome is caused by a mutation in the gene encoding the cardiac sodium channel (SCN5A) and is common in Southeastern Asia, where it is the second highest cause of death among young men.72 In Europe, gender specificity is less distinct, although affected women tend to be less symptomatic than affected men. Patients with Brugada syndrome often die in their sleep, and a possible relationship to sudden infant death syndrome has been suggested.73
It is believed that the mechanism of VF in patients with Brugada syndrome is related to reduced function of the mutant late inward sodium current, forcing the balance of plateau currents in favor of repolarization. This is why any further impairment of sodium channel function (e.g., by sodium-blocking drugs) can exacerbate the electrocardiographic and arrhythmogenic phenotype in this disorder.74 Because epicardial, not endocardial, cells possess the transient outward current (Ito), they are most susceptible to the repolarizing effect of the sodium channel mutation in Brugada syndrome. Marked and selective shortening of epicardial action potentials relative to endocardial action potentials produces a transmural voltage gradient during the plateau that, in turn, is believed to account for the characteristic pattern of ST segment elevation seen in these patients.75 Transmural action potential gradients also account for the presumed mechanism of VF (i.e., phase 2 re-entry).
In a recent provocative study, Haisaguerre et al identified a select group of patients with recurrent idiopathic VF that seemed to originate with premature beats emanating from the distal His-Pukinje system; these patients were successfully treated with radiofrequency ablation targeting the originating premature beats, with a low rate of VF recurrence.76
Clinical Electrocardiography
The role of electrocardiography has been increasing in patients presenting with VF. Certain ECG features may identify high-risk patients or transient events that can result in VF, which may be preceded by VT (see Chapter 47).
Electrocardiographic Features of Patients at High Risk for Ventricular Fibrillation
Electrocardiographic recordings obtained at the onset of VF provide insight into events that precipitate SCD. Documentation of these events has been obtained by continuous monitoring in hospital telemetry units and by ambulatory monitoring. The patients involved in these studies had a high incidence of CAD, and most had frequent or complex ventricular ectopy. The terminal events were associated with sinus arrest, complete heart block, or ventricular asystole in approximately 10% of patients; in 90%, VF was preceded by VT or ventricular flutter of variable duration.77–80 ST segment and T-wave changes indicative of ischemia related to acute MI or coronary spasm have also been reported to precede terminal ventricular arrhythmias.77–80 Other studies indicate that bradycardia and electromechanical dissociation are important causes of SCD in patients with advanced heart failure and nonischemic cardiomyopathy.81 These observations have significant implications for strategies to reduce mortality rates in patients at risk for SCD.
The diagnostic role of the ECG in identifying patients with genetic disorders associated with SCD from ventricular arrhythmias has continued to evolve over the past decade. The electrocardiographic characteristics of LQTS, Brugada syndrome, and arrhythmogenic right ventricular cardiomyopathy (ARVC) have gained widespread attention. This is vital to the prevention of SCD in young patients with these disorders, particularly if these patients participate in athletics. A recent study has suggested a higher incidence of early repolarization changes in the inferolateral leads (J-point elevation and notching in the terminal portion of the QRS complex) among survivors of cardiac arrest caused by idiopathic VF when compared with control subjects.82
The pattern of early repolarization associated with highest risk seems to be global or inferolateral early repolarization with prominent J waves on the resting ECG.83 It is not clear if a single disorder of repolarization causes at least some of the cases previously labeled “idiopathic VF,” or, as seems more likely, a spectrum of disorders is associated with abnormal and heterogeneous repolarization that predisposes otherwise normal individuals to VF. These disorders have been collectively named early repolarization disease.84
Long QT Syndrome
The electrocardiographic manifestations of LQTS include QT prolongation, abnormalities in T-wave morphology, increases in QT dispersion, T-wave alternans, and a relative degree of bradycardia in children (Figure 46-11). The upper limits of normal for the QTc values are 460 to 470 ms for females and 440 to 460 ms for males.85,86 Longer QT values may be observed in normal women after puberty.87 The degree of QT prolongation does not directly correspond with the risk of syncope, but malignant ventricular arrhythmias are more frequent when the QTc exceeds 600 ms.87 A diagnostic dilemma is that the Q-T interval shows temporal variations in patients with this syndrome, and the QTc may fall within the normal range on a random recording.88 Garson reported that 6% of LQTS patients had a normal Q-T interval, and data from the International Registry showed that 10% of family members with a QTc less than 440 ms had a cardiac arrest.89,90 Thus, an ECG with a normal QTc does not exclude the diagnosis if strong suspicion exists that a patient has the syndrome, especially if the QTc is on the border of normal.
The effect of exercise increases the QTc in patients with LQTS, but this effect is less apparent in patients with LQT3 than in those with other genotypes.91 Approximately 62% of patients with LQTS exhibit T waves that are biphasic or notched, and a higher incidence of these abnormalities is seen in patients with cardiac events.92 The characteristic features are most pronounced in precordial leads V2 to V5. The appearance of notched T waves may be provoked by exercise. The degree of QT dispersion is measured by the difference between the longest and shortest Q-T intervals on the 12-lead ECG and is prolonged in patients with LQTS.93 It is thought to represent increased dispersion of repolarization. Patients who show no change in the degree of QT dispersion when they are treated with β-blockers appear to be at increased risk for cardiac events.90,94 T-Wave alternans is a beat-to-beat alternation in the amplitude or polarity of the T wave. It appears to be a marker of electrical instability that may precede torsades de pointes.94 Children with LQTS often have resting heart rates that are lower than normal and may exhibit a blunted chronotropic response to exercise.87,95 Torsades de pointes, which is the ventricular arrhythmia associated with LQTS, is characterized by the undulating amplitude of the QRS complex, which gives the appearance of twisting about its axis. The onset is frequently associated with pause-dependent ventricular ectopy that falls on the T wave.96–98
Brugada Syndrome
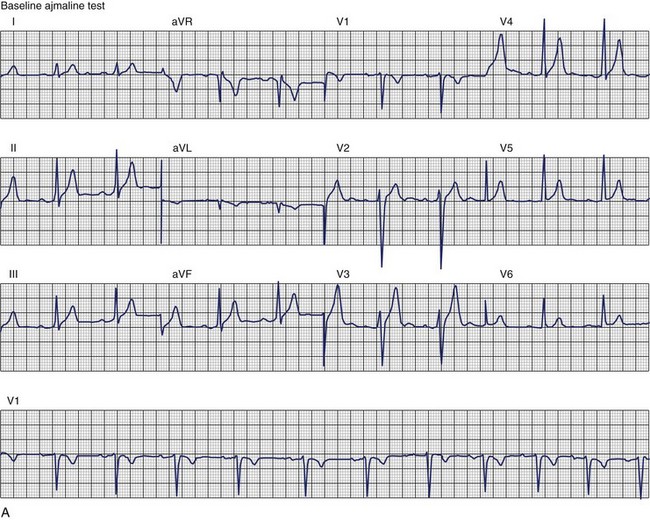
The ECG patterns associated with Brugada syndrome are (1) a terminal R′ in lead V1 with complete or incomplete RBBB; (2) convex downward (coved) ST segment elevation equal to 0.1 mV in lead V1 or leads V1 and V2; convex upward (saddle-shaped) ST-segment elevation equal to 0.1 mV; and (3) J-point elevation followed by a downsloping ST segment ending in a negative deflection (triangular shape).98 Serial ECGs performed on the same patient may show variation from one pattern of ST-segment elevation to another, normalization, and progressive development of RBBB. Figure 46-12 shows examples of variable Brugada ECG patterns that occurred during an ajmaline test in the same patient.99,100 The prevalence of the Brugada ECG pattern is reported to be 0.07% to 0.7%, and there is a male predominance that is especially marked in Asians.101–106 The range in prevalence appears to depend on the criteria used to make the diagnosis. The saddleback ST-segment elevation is more common. The typical coved pattern was found in 0.1% to 0.26% of community-based populations in Japan and Europe.104–106 In a Japanese study population that underwent ECGs during health examinations, the prevalence of all types of Brugada ECG patterns was 0.7%.103 The coved-type ST-segment elevation was found in 38% of subjects with the Brugada pattern, and the saddleback-type ST-segment elevation was seen in 62%. In the same study, the rsR′ pattern in lead V1 was observed in 41%, and the Rsr′ pattern was recorded in 59%. The prevalence of the coved pattern was 0.26%, and the typical Brugada ECG pattern with coved ST-segment elevation and the rsR′ pattern in lead V1 was 0.12%. If only male subjects were considered, the criteria for a Brugada ECG pattern was met in 2.14% of the population, but the typical pattern in males was 0.38%.104 In a European population studied by Hermida, the prevalence of ST segment elevation was 6.1%; however, only 1 (0.1%) of the 61 subjects who met the study’s criteria for the Brugada pattern had the coved pattern. All of the others had the saddleback pattern.103
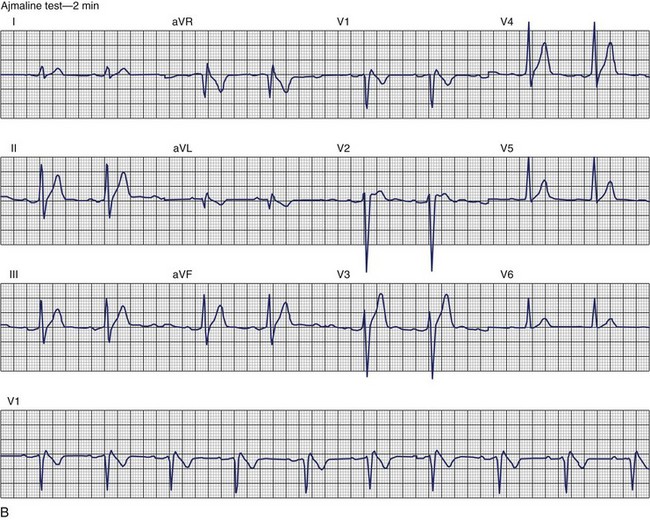
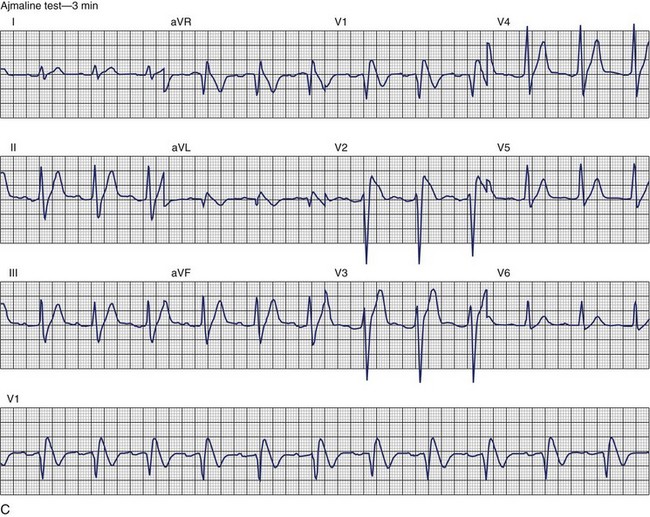
The prognostic significance of the Brugada ECG pattern is difficult to assess. Brugada reported an 8% incidence of arrhythmic events in an asymptomatic hospital-based population.106 The degree and type of ST segment elevation require further study for risk stratification of asymptomatic individuals in community-based populations. Matsuo evaluated mortality in patients younger than 50 years who had ECGs recorded during biannual examinations from 1958 through 1999.107 A total of 32 patients were identified with the Brugada ECG pattern. Seven of these patients died suddenly or died of an unexplained accident. Although total mortality was not increased in patients with the Brugada ECG pattern, the mortality rate from unexpected death was significantly higher. No increase in mortality was observed in studies by Miyasaka, Takenaka, or Priori.104,105,107 In the Osaka population, one SCD occurred among the 98 subjects with the Brugada ECG pattern during a mean follow-up of 2.6 years.104 A 3-year follow-up reported by Atarashi of patients with a Brugada ECG pattern found cardiac event–free rates of 67.6% in symptomatic patients and 93.4% in an asymptomatic group.108 Coved-typed ST segment elevation appeared to be related to cardiac events. The higher incidence in hospital-based studies may reflect referral patterns based on a family history of SCD. Differences in criteria or ECG interpretation affect the diagnosis of this pattern, and the follow-up in most studies is too short to draw definitive conclusions about the long-term prognosis.
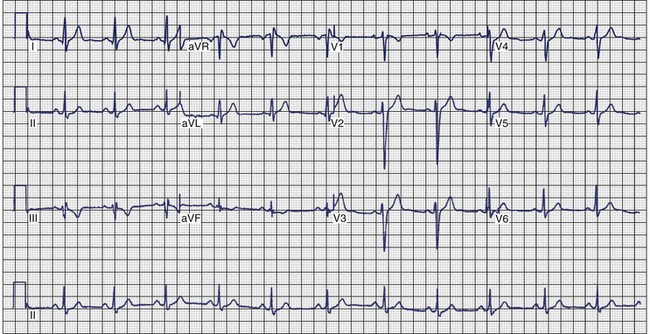
Arrhythmogenic Right Ventricular Cardiomyopathy
ECG recordings during sinus rhythm in patients with ARVC have several distinctive features (Figure 46-13).109 The QRS may be prolonged in the right precordial leads to a greater extent than in leads I or V6. The QRS is often greater than 110 ms in lead V1 (sensitivity, 55%), and a pattern of incomplete RBBB is observed.110 In 30% of cases, the delay in conduction over the right ventricle results in a small potential at the terminal portion of the QRS in lead V1 that has been termed an epsilon (ε) wave, which can be amplified with bipolar recordings over the inferior and superior aspects of the sternum.109 This can be achieved by repositioning the left arm lead over the xiphoid process, positioning the right arm lead over the manubrium sternum, and applying the left leg lead at the position customarily used for lead V4 or V5.111 The other major feature of ECGs recorded from patients with ARVC is inversion of the T waves in the precordial leads, which is observed in 42% to 54% of patients.111,112 Metzger assessed the value of serial 12-lead ECGs in 20 patients to recognize progression of ARVC over a mean of 71 ± 48 months.113 Abnormalities were detected in 90% of the patients. The most frequent abnormality was T-wave inversion in the precordial leads. No correlation was demonstrated between the ECG and the extent of disease detected by echocardiography. In the 14 patients who had several ECGs recorded over time, no clear progression of electrocardiographic abnormalities was observed.
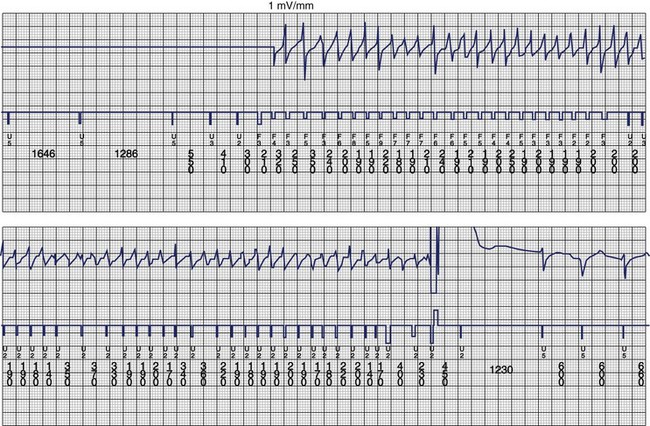
The ventricular arrhythmias associated with ARVC typically show a morphology resembling left bundle branch block in lead V1 with a variable axis. Nava published a clinical profile and long-term follow-up of 37 families with ARVC that demonstrated a correlation between the severity of echocardiographic findings and the severity of ventricular arrhythmias, which were seen in all patients with the severe form of the disease, in 82% with moderate disease, and in 23% in those with mild disease.112 Overall, 60 (45%) of 132 affected living members had ventricular arrhythmias. These included VF in 1, sustained VT in 14, nonsustained VT in 8, ventricular couplets and triplets in 16, and frequent ventricular premature depolarizations in 8. Exercise-induced polymorphic VT was observed in 13 patients with no other documented arrhythmias. Only one patient who was judged to have mild disease had a sustained ventricular arrhythmia. Although the data from this study showed a low incidence of VF, the incidence may be higher. In 19 of the 37 families, the proband died at a young age, and the diagnosis was made at autopsy. One may speculate that some of these subjects had VF. Figure 46-14 shows electrograms recorded from an ICD implanted in a teenager (male) with ARVC and frequent nonsustained ventricular arrhythmias. The recording shows the sudden onset and successful termination of VF, which occurred at night while he was asleep. He had no prior history of syncope or sustained ventricular arrhythmias.
Diagnostic Evaluation
Evaluation of Transient or Reversible Causes
VF that occurs secondary to a reversible or transient cause may be adequately treated by correction of the reversible cause or by short-term therapy or close observation while awaiting spontaneous resolution of the transient cause.114 The causes of VF with these characteristics are usually readily identified with a few focused investigations. Electrolyte abnormalities are identified with serum electrolyte testing performed as soon as possible after resuscitation. The most common electrolyte abnormalities leading to VF are hypokalemia, hypomagnesemia, or both. When considering the temptation to ascribe an episode of VF wholly to hypokalemia, hypomagnesemia, or both, one must recall that the adrenergic discharge state during resuscitated VF results in redistribution of extracellular potassium and magnesium into the intracellular compartment. Accordingly, relative hypokalemia, hypomagnesemia, or a combination is very common after VF.115 Only marked hypokalemia, hypomagnesemia, or both and confidence that future episodes can be prevented should prompt the belief that the episode of VF had a reversible cause.
The early performance of a 12-lead ECG and serum markers of myocardial necrosis (creatine kinase [muscle or brain type], troponin) will permit identification of the patient whose VF has occurred in the acute phase (first 48 hours) of an MI. Evidence that acute MI produces an environment that constitutes only a transient risk of VF is most convincing for a Q-wave MI.116 Nevertheless, the risk of VF may also be transient in the setting of a non–Q-wave MI.117 However, the practitioner must avoid the temptation to ascribe an episode of VF wholly to an acute MI if the only evidence of acute myocardial necrosis is a marginal elevation in a serum marker of necrosis; such elevations may be secondary to the VF-induced cardiac arrest rather than represent its cause. The diagnosis of MI in the setting of out-of-hospital cardiac arrest from VF is made difficult by the usual absence of a history of chest pain, frequent nondiagnostic ECG abnormalities that may be a consequence of the myocardial ischemia resulting from the global “no-flow or low-flow state” during VF, and the absence of a specific cut-off for the degree of cardiac enzyme elevation that separates MI causing VF from VF causing myocardial necrosis. In general, the presence of a culprit lesion (coronary thrombus or ulcerated plaque) on angiography early after cardiac arrest and well-preserved or normal left ventricular function suggests an ischemic or infarction-related cause.
The other common reversible or transient cause of VF is the use of a proarrhythmic drug: a classic antiarrhythmic drug, other drugs with electrophysiological effects, and recreational drugs, particularly cocaine.118 Use of these agents is determined from the history. However, if the practitioner believes that a report of such drug use will not be forthcoming from a high-risk individual, a toxicology screen is advised. When therapeutic drug use is reported, early determination of a serum concentration of the agent may be important when toxicity related to that agent has a relationship to serum concentrations (i.e., digitalis).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


