20
Vascular anomalies
Introduction
Vascular anomalies are common in infants and children, but their classification is frequently a source of great confusion and, as a result, they are often poorly managed. This is despite the fact that, as long ago as 1982, Mulliken and Glowacki proposed a new way of classifying vascular anomalies1 that is now the most widely recognised system in use. It is based upon differences in the cellular kinetics and natural history of these lesions and describes two separate groups: vascular tumours and vascular malformations, which will be discussed in turn. Vascular tumours include the common infantile haemangiomata as well as rarer congenital haemangiomata and other vascular tumours, many of which are high flow. Vascular malformations comprise low-flow venous and lymphatic malformations and high-flow arteriovenous malformations. An acquired arteriovenous fistula following trauma is a separate entity that may occasionally be misdiagnosed as a vascular malformation.
Vascular tumours
Infantile haemangioma
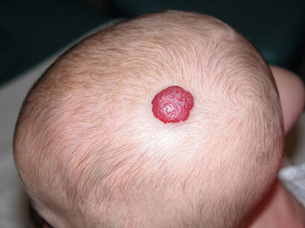
Typically, this lesion is not present on the day of birth but becomes visible within the first 2 months of life. An initial stage of rapid proliferation first brings the lesion to attention, although the clinical appearance of the haemangioma at this time will depend upon its depth of involvement of the skin. Those involving the superficial dermis produce lobulated, bright red lesions that are commonly referred to as ‘strawberry birthmarks’ (Fig. 20.1). A haemangioma involving the deep dermis and subcutis, however, will produce a swelling with either no discoloration or a blueness of the overlying normal skin. The term ‘cavernous haemangioma’ is often used to describe such lesions, but it is important to recognise that their appearance is due solely to their depth of involvement of the skin and not to any variation in their histology.
PHACE syndrome2
Management of haemangiomata of infancy
The majority of haemangiomata can be managed conservatively but intervention is indicated when a lesion causes significant mass effect or disfigurement, when it involves the airway and when it obstructs the visual axis. Pharmacological management is now the mainstay of treatment in this group, with beta-blockers the preferred first-line therapy.3 Rarely, high output cardiac failure occurs in children with very large infantile haemangiomata, particularly when there is hepatic involvement, and this requires urgent intervention. Management consists of a combination of optimal medical therapy and particle embolisation of the capillary bed, with the aim of reducing the vascularity of the lesion and stabilising the child’s cardiovascular status until the natural history of the lesion causes involution.
Other vascular tumours
KMP describes a pattern of variable but often severe coagulopathy and thrombocytopenia seen in association with a variety of soft-tissue lesions, most commonly KHE, and is associated with a high mortality. It is not a complication of the common haemangioma of infancy5 and should not be confused with the coagulopathy that may be associated with large venous malformations in which there is consumption of clotting factors with elevated D-dimers but a much less profound reduction in platelet and fibrinogen levels. The management of this condition lies outside the scope of this chapter but evolving pharmacological approaches to other vascular tumours may have a role to play in the management of these lesions and embolisation may be helpful in some cases.
Vascular malformations
Low-flow malformations
Capillary malformations come in a variety of forms.
Salmon patch (naevus simplex; erythema nuchae): A salmon patch is a red macule present at birth, which most commonly involves the skin of the nape of the neck, the upper eyelids or glabella. It is usually central, does not follow a dermatomal distribution and will usually fade by 2 years of age, especially if it involves the skin of the face; the nuchal lesion is more likely to persist into adult life.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


