The development of acute pulmonary edema involves a complex interplay between the capillary hydrostatic, interstitial hydrostatic, and oncotic pressures and the capillary permeability. We review the pathophysiological processes involved and illustrate the concepts in a number of common clinical situations including heart failure with normal and reduced ejection fractions, mitral regurgitation, and arrhythmias. We also describe other rarer causes including exercise, swimming, and diving-induced acute pulmonary edema. We suggest a unifying framework in which the critical abnormality is a mismatch or imbalance between the right and left ventricular stroke volumes. In conclusion, we hypothesize that increased right ventricular contraction is an important contributor to the sudden increase in capillary hydrostatic pressure, and therefore, a central mechanism involved in the development of alveolar edema.
The purpose of this review is to consider new insights into the mechanisms involved in the development of acute pulmonary (alveolar) edema secondary to hemodynamic abnormalities and describe the possible pathophysiological processes involved in both common and rare causes. Pulmonary edema because of adult respiratory distress syndrome and sepsis is beyond the scope of this review. Acute pulmonary edema (APE) is a dramatic medical emergency. The pulmonary airspaces fill with liquid, and the patients start to drown in their own fluids. The pathophysiology is usually described in terms of a failing left ventricle requiring an ever-higher filling pressure to function: the consequent increase in pulmonary capillary hydrostatic pressure because of backward pressure causes transudation of fluid from the capillaries into the airspaces. However, as de Bono eloquently wrote “it is salutary to be reminded that patients with disease are often the most appropriate model for studying human pathophysiology, and when clinical observations do not tally with theories, it is usually the latter that are wrong.” One such observation is that pulmonary edema may occur in subjects with an apparently normal heart given the right circumstances.
Pulmonary Microcirculation Anatomy and Physiology
The walls of the alveoli consist of type I and type II epithelial cells (pneumocytes; Figure 1 ). Type I cells form 90% of the alveolar cell surface area and are fragile. Type II cells are more robust, produce surfactant, transport ions, and regulate fluid flow out of the alveoli. Type II cells can also proliferate and differentiate into type I cells. Fluid and solute filtration occur across the pulmonary capillary endothelium into the adjacent interstitial space. Gas exchange takes place in the thin segment between the capillary and the alveolar wall ( Figure 1 ). At this point, the endothelial and epithelial cells’ basement membranes (basal lamina) become tightly fused into a single layer. The total capillary blood volume in the lungs is ∼70 ml (∼10% of the pulmonary circulation volume) and is similar to the right ventricular stroke volume. Approximately 1/3 of the pulmonary vascular resistance is because of the pulmonary capillaries. Capillary hydrostatic pressure (Although the term hydrostatic pressure is widely used in clinical practice and physiology literature, the term should strictly only be applied to stationary fluids. A more accurate term is the hydrodynamic lateral pressure and differs from the pressure measured along the direction of travel of the fluid.) is usually closely determined by pulmonary artery pressure because of the relatively low pulmonary vascular resistance.
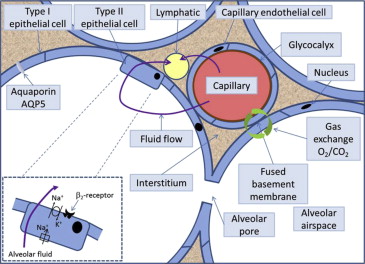
The interstitial fluid moves toward the hilum along the spaces beside the vessels and the airways. The excess filtrate is removed by the pulmonary lymphatic system. Lymphatic flow arises because the interstitial hydrostatic pressure is more negative closer to the hilum; flow is assisted by the cyclic external compression that occurs during the breathing cycle coupled with the presence of 1-way valves in the lymphatic vessels and intrinsic lymphatic peristaltic contraction. The lymphatic vessels drain into the systemic venous system through the thoracic duct. The lymphatic channels can dilate in chronic situations, such as mitral stenosis, suggesting that an adaptive increase in flow at rest can occur.
Starling’s Hypothesis Revisited
In 1896, Starling described the relation between the fluid flow across the capillary membrane (flux) and the hydrostatic and oncotic pressures ( Figure 2 ). The Starling equation states that the net filtration (J v ) is proportional to the net sum of hydrostatic and oncotic pressures. Levick and Michel have recommended some modifications to the basic equation, where Π g (oncotic pressure immediately beneath the glycocalyx) replaces Π c . By convention, an outward “force” (or, more correctly, pressure) is positive and an inward one is negative. Capillary hydrostatic pressure drives the fluid out of the vasculature and is opposed by the interstitial hydrostatic pressure.
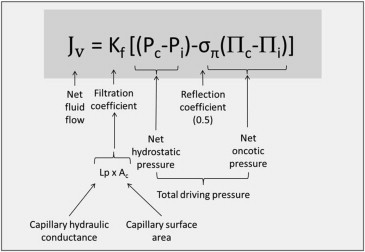
Hydrostatic pressures (P) are usually measured in millimeter of mercury. The filtration coefficient (K f ) is a measure of ease of fluid (solvent or water) movement across the alveolar membrane. In humans, the lungs’ total K f is estimated to be 10 ml/min/cmH 2 O and is lower than most other organs. The capillary hydrostatic pressure (P c ) is normally ∼13 mm Hg at arteriolar end and 6 mm Hg at venous end. The interstitial hydrostatic pressure (P i ) is approximately −2 mm Hg relative to atmospheric pressure. The net hydrostatic pressure (ΔP) is the difference between capillary and interstitial pressures and varies between the upper and lower lungs because of the difference in height and the effect of gravity (determined by ρgh, where ρ, g, and h denotes fluid density, gravitational constant, and height, respectively). A difference in height between the apex and base of the lung is ∼25 cm. There is, thus, a pressure difference of 25 cmH 2 O (18 mm Hg) between the apex and base of the lungs while standing. A typical pulmonary artery pressure is 25/8 mm Hg, sufficient for perfusion of the highest part of the lung while upright.
Oncotic pressure is the colloid osmotic pressure generated by colloidal solute components and is an inward pressure ( Figure 3 ). The interstitial oncotic pressure is high because of a leak of protein (mostly albumin) across the thin capillary membrane. The pulmonary capillaries have a reflection coefficient (σ π ) of ∼0.5. Normally, the hydrostatic pressure exceeds the opposing oncotic pressure along the capillary’s full length, and so capillaries are continuously filtering ( Figure 3 ). The small net outward movement of fluid from the pulmonary capillaries is estimated to be 0.3 ml/min in a 70-kg human.
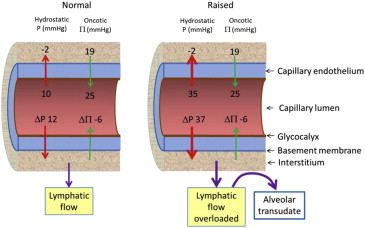
For pulmonary edema to occur, excess fluid must first form in the interstitium before flowing into the alveoli. It is caused by an imbalance in Starling “forces”. The 3 most common causes are an increase in capillary hydrostatic pressure (P c ), an increase in capillary permeability (K f ), and an inadequate clearance by the pulmonary lymphatic system. For example, when there is increased capillary hydrostatic pressure (P c ), more fluid is driven out of the circulation and into the interstitium ( Figure 4 ). A decrease in hydrostatic pressure in the interstitium (P i ) from rapid evacuation of pleural fluid, pneumothorax, and acute upper airway obstruction results in a lowering of P i (hence increasing ΔP) causing an increase in net fluid flux (J v ). Pure hydrostatic pulmonary edema is a transudate without inflammatory cells.
Physiological mechanisms have evolved to resist the development of pulmonary edema. These are (a) increased lymph flow, (b) decreased interstitial oncotic pressure, and (c) high interstitial compliance.
- (a)
An increased fluid filtration (F v ) causes an increase in interstitial hydrostatic pressure and lymph flow that tend to remove the fluid. However, because the lymphatic vessels drain into the systemic veins, the flow may be countered by an increase in venous pressure.
- (b)
Increased lymph flow washes the albumen out of the interstitium lowering the interstitial oncotic pressure (oncotic buffering). The combination of increased lymph flow and decreased albumen concentration reduces interstitial oncotic pressure, thus increasing the oncotic pressure gradient favoring retention of fluid in the capillary.
- (c)
A large volume of fluid (300 to 400 ml in an adult) can be accommodated in the gel of the interstitium without an increase in interstitial pressure because of the high interstitial tissue compliance.
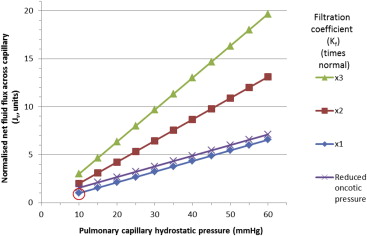
Once the interstitium is full, fluid overflows into the alveoli. The capillary hydrostatic pressure must increase 3-fold before alveolar flooding occurs (equivalent to a pulmonary capillary wedge pressure of 25 to 30 mm Hg) in a dog model using aortic constriction ( Figure 4 ). The protective mechanisms are largely ineffective if the capillary membrane is damaged through inflammatory, allergic, or septic conditions. However, secondary capillary damage is probably common with high hemodynamic pressures. The high pressure causes direct insult through increasing shear stress and by damaging both the glycocalyx lining and the endothelium ( Figures 1 and 3 ). Injury to the lung capillaries causes them to leak both solutes and protein so that the resulting edema fluid is an exudate.
When albumin levels in blood are low, as happens in nephrotic syndrome or liver disease, the lower capillary oncotic pressure increases the flux of fluid forced into the interstitial tissues. The protein content of the interstitium is low, and thus, the edema fluid is a transudate. An isolated very low capillary oncotic pressure has only a limited effect on fluid flux ( Figure 4 ), but when a low albumin is combined with a modest increase in hydrostatic pressure, it can result in edema formation ( Figure 4 ). There is active reabsorption of fluid from the alveoli driven by sodium transport pumps ( Figure 1 ). AQP5 (aquaporin type 5) in the membrane of aquaporin water channels in type I epithelial cells contribute to water permeability although their role in pulmonary edema is uncertain.
Starling’s Hypothesis Revisited
In 1896, Starling described the relation between the fluid flow across the capillary membrane (flux) and the hydrostatic and oncotic pressures ( Figure 2 ). The Starling equation states that the net filtration (J v ) is proportional to the net sum of hydrostatic and oncotic pressures. Levick and Michel have recommended some modifications to the basic equation, where Π g (oncotic pressure immediately beneath the glycocalyx) replaces Π c . By convention, an outward “force” (or, more correctly, pressure) is positive and an inward one is negative. Capillary hydrostatic pressure drives the fluid out of the vasculature and is opposed by the interstitial hydrostatic pressure.
Hydrostatic pressures (P) are usually measured in millimeter of mercury. The filtration coefficient (K f ) is a measure of ease of fluid (solvent or water) movement across the alveolar membrane. In humans, the lungs’ total K f is estimated to be 10 ml/min/cmH 2 O and is lower than most other organs. The capillary hydrostatic pressure (P c ) is normally ∼13 mm Hg at arteriolar end and 6 mm Hg at venous end. The interstitial hydrostatic pressure (P i ) is approximately −2 mm Hg relative to atmospheric pressure. The net hydrostatic pressure (ΔP) is the difference between capillary and interstitial pressures and varies between the upper and lower lungs because of the difference in height and the effect of gravity (determined by ρgh, where ρ, g, and h denotes fluid density, gravitational constant, and height, respectively). A difference in height between the apex and base of the lung is ∼25 cm. There is, thus, a pressure difference of 25 cmH 2 O (18 mm Hg) between the apex and base of the lungs while standing. A typical pulmonary artery pressure is 25/8 mm Hg, sufficient for perfusion of the highest part of the lung while upright.
Oncotic pressure is the colloid osmotic pressure generated by colloidal solute components and is an inward pressure ( Figure 3 ). The interstitial oncotic pressure is high because of a leak of protein (mostly albumin) across the thin capillary membrane. The pulmonary capillaries have a reflection coefficient (σ π ) of ∼0.5. Normally, the hydrostatic pressure exceeds the opposing oncotic pressure along the capillary’s full length, and so capillaries are continuously filtering ( Figure 3 ). The small net outward movement of fluid from the pulmonary capillaries is estimated to be 0.3 ml/min in a 70-kg human.
For pulmonary edema to occur, excess fluid must first form in the interstitium before flowing into the alveoli. It is caused by an imbalance in Starling “forces”. The 3 most common causes are an increase in capillary hydrostatic pressure (P c ), an increase in capillary permeability (K f ), and an inadequate clearance by the pulmonary lymphatic system. For example, when there is increased capillary hydrostatic pressure (P c ), more fluid is driven out of the circulation and into the interstitium ( Figure 4 ). A decrease in hydrostatic pressure in the interstitium (P i ) from rapid evacuation of pleural fluid, pneumothorax, and acute upper airway obstruction results in a lowering of P i (hence increasing ΔP) causing an increase in net fluid flux (J v ). Pure hydrostatic pulmonary edema is a transudate without inflammatory cells.
Physiological mechanisms have evolved to resist the development of pulmonary edema. These are (a) increased lymph flow, (b) decreased interstitial oncotic pressure, and (c) high interstitial compliance.
- (a)
An increased fluid filtration (F v ) causes an increase in interstitial hydrostatic pressure and lymph flow that tend to remove the fluid. However, because the lymphatic vessels drain into the systemic veins, the flow may be countered by an increase in venous pressure.
- (b)
Increased lymph flow washes the albumen out of the interstitium lowering the interstitial oncotic pressure (oncotic buffering). The combination of increased lymph flow and decreased albumen concentration reduces interstitial oncotic pressure, thus increasing the oncotic pressure gradient favoring retention of fluid in the capillary.
- (c)
A large volume of fluid (300 to 400 ml in an adult) can be accommodated in the gel of the interstitium without an increase in interstitial pressure because of the high interstitial tissue compliance.
Once the interstitium is full, fluid overflows into the alveoli. The capillary hydrostatic pressure must increase 3-fold before alveolar flooding occurs (equivalent to a pulmonary capillary wedge pressure of 25 to 30 mm Hg) in a dog model using aortic constriction ( Figure 4 ). The protective mechanisms are largely ineffective if the capillary membrane is damaged through inflammatory, allergic, or septic conditions. However, secondary capillary damage is probably common with high hemodynamic pressures. The high pressure causes direct insult through increasing shear stress and by damaging both the glycocalyx lining and the endothelium ( Figures 1 and 3 ). Injury to the lung capillaries causes them to leak both solutes and protein so that the resulting edema fluid is an exudate.
When albumin levels in blood are low, as happens in nephrotic syndrome or liver disease, the lower capillary oncotic pressure increases the flux of fluid forced into the interstitial tissues. The protein content of the interstitium is low, and thus, the edema fluid is a transudate. An isolated very low capillary oncotic pressure has only a limited effect on fluid flux ( Figure 4 ), but when a low albumin is combined with a modest increase in hydrostatic pressure, it can result in edema formation ( Figure 4 ). There is active reabsorption of fluid from the alveoli driven by sodium transport pumps ( Figure 1 ). AQP5 (aquaporin type 5) in the membrane of aquaporin water channels in type I epithelial cells contribute to water permeability although their role in pulmonary edema is uncertain.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


