The Lungs in Patients with Inborn Errors of Metabolism
INTRODUCTION
Inborn errors of metabolism, also termed “inherited metabolic disorders,” are a group of inherited systemic conditions involving various types of chemical imbalances that cause disease and affect essentially all organs to a greater or lesser degree.1–9 Each of the diseases is characterized by a specific genetic defect that causes an abnormality of an enzyme necessary for the degradation of some chemical substance.2 The disruption of degradation leads to disease due to the deficiency of a substance or to the pathologic accumulation of a substance.2,5 The first inborn errors of metabolism were identified about a century ago. Sir Archibald Garrod coined the phrase in 1902.10 Today, there are over 500 inborn errors of metabolism identified.11
The diseases that comprise the inborn errors of metabolism are individually rare; however, taken together, they affect about 1 in 1000 people.2 The majority of the diseases making up the inborn errors of metabolism exhibit autosomal recessive inheritance; however, X-linked recessive inheritance occurs with some diseases, and, less commonly, autosomal dominant inheritance is seen.5 The majority of these diseases are pediatric; however, with the identification of attenuated variants, and with improved survival, they are today’s conditions that must be considered in patients of all ages.2 Tissue biopsy may be necessary for specific diagnosis; however, given that these disorders typically present with unexpected findings, high clinical suspicion and careful radiologic evaluation are usually the key to faster and more accurate diagnoses. Particularly as newer and more effective therapies being designed require earlier diagnoses, radiology plays a central role in diagnosis, and clinical–radiologic correlations are increasingly important.2 Although metabolic specialists with expertise in the various inborn errors of metabolism are present in large academic centers and tertiary care centers, pediatricians and primary care physicians are typically the first physicians to whom patients with these diseases present themselves.5
Some of the diseases that make up the inborn errors of metabolism can be placed into a few broad groups; these include disorders of amino acid metabolism, organic acidurias, urea cycle defects, disorders of ketogenesis and ketolysis, disorders of fatty acid oxidation, lysosomal storage disorders, and mitochondrial disease.2 While the lung is not typically a primary site of clinical disease in patients with inborn errors of metabolism, the lungs are involved with many inborn errors of metabolism, and in some diseases, clinically significant lung disease may occur.1,12 Of course, the lungs may be involved with infections as a secondary feature in many types of chronic diseases, including inborn errors of metabolism; however, this chapter focuses on several specific inborn errors of metabolism for which lung disease may be a clinically significant feature.
ACID SPHINGOMYELINASE DEFICIENCY (NIEMANN–PICK DISEASE TYPES A AND B)
Acid sphingomyelinase (ASM) deficiency, an autosomal recessive condition, has a phenotype that occurs in a continuum, with severe, neuropathic disease with early symptoms and resulting in death in infancy or early childhood designated as Niemann–Pick disease type A (NPA), and nonneuropathic disease with generally later onset and milder symptoms designated as Niemann–Pick disease type B (NPB).13–17 Patients with NPA typically show hepatosplenomegaly by about 3 months of age. The hepatosplenomegaly may ultimately become massive, and children are often dead within 3 years. Psychomotor development in patients with NPA typically is limited to not more than the 12-month level, after which deterioration occurs. NPB patients generally show milder hepatosplenomegaly and may survive to adulthood; however, progressive and clinically significant pulmonary changes are frequent in these patients.14,18,19 Diagnosis is made by showing less than 10% normal residual ASM activity in peripheral blood lymphocytes or cultured skin fibroblasts.
Patients with NPA often exhibit radiographic changes of interstitial lung disease due to sphingomyelin storage within pulmonary macrophages, and they may exhibit low arterial blood oxygen levels. Respiratory infections are common, and not infrequently culminate in respiratory failure and death.13,20,21 Patients with NPB, who have generally milder disease, may develop pulmonary complications at any age.21,22 Most patients with NPB exhibit radiographic changes of interstitial lung disease, even while their individual symptoms may vary dramatically; their radiographic changes often do not correlate with the severity of pulmonary function abnormalities.14,18 Cystic lung disease has also been reported in association with NPB.23 Whole lung lavage has been attempted for symptomatic treatment of patients with NPB.24
Radiographically, patients show nodular infiltrates and linear strands, with a predominantly basilar honeycomb pattern.19,25 Histologically, the lungs demonstrate predominantly normal architecture, with airspaces filled with Niemann–Pick cells—enlarged histiocytes with finely vacuolated cytoplasm (Figs. 67-1 and 67-2).26 If necessary, CD68 immunostain may be used to establish their presence. These collections of cells have been termed “sea blue histiocytosis,” due to the vivid blue staining with May–Grunwald Giemsa stain. Pulmonary fibrosis may occur over time (Fig. 67-3).
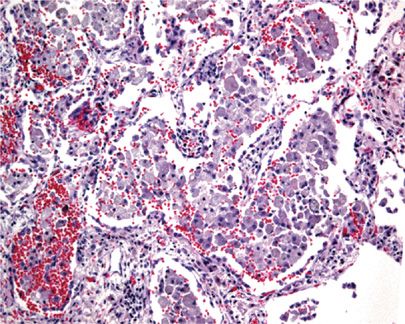
Figure 67-1 Low power image of lung in Niemann–Pick disease showing collections of foam cells within alveoli.
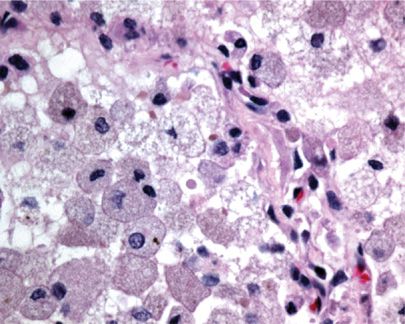
Figure 67-2 High power image of lung in Niemann–Pick disease showing enlarged histiocytes with finely vacuolated cytoplasm within the alveolar spaces.
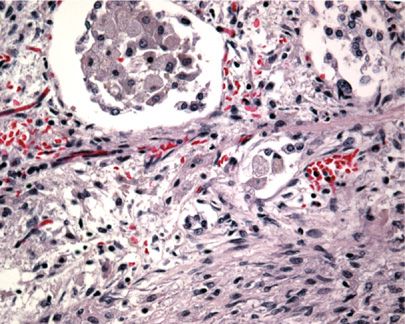
Figure 67-3 High power image of lung in long-standing Niemann–Pick disease showing developing pulmonary fibrosis.
NIEMANN–PICK DISEASE TYPE C
Niemann–Pick disease type C (NPC) is a rare, autosomal recessive inborn error of metabolism characterized by impaired intracellular lipid trafficking and accumulation of cholesterol and glycosphingolipids in the brain and other tissues.27–34 It is diagnosed by biochemical tests that show impaired cholesterol esterification and positive filipin staining in cultured fibroblasts. Patients typically have saccadic eye movement abnormalities or vertical supranuclear gaze palsy; cerebellar signs, such as ataxia, dystonia/dysmetria, dysarthria, and dysphagia; gelastic cataplexy; and epileptic seizures27,33 are given symptomatic treatment for cataplexy, dystonia, and seizures, as well as physical therapy to maintain as much independent mobility as possible. Approximately 95% of NPC cases are due to mutations of the NPC1 gene, with the remainder due to mutations of the NPC2 gene.27
NPC may produce symptoms in patients of any age; and initial presentation may occur from neonatal to adult periods. Infants often have nonspecific symptoms that make exact diagnosis difficult. Along with liver disease, infants may present with often-fatal pulmonary failure due to impaired gas exchange due to lung infiltration with foam cells. Patients presenting in childhood or adolescence, or as adults, typically do not have pronounced pulmonary involvement.30–32,35 Pulmonary alveolar proteinosis may occur in patients with NPC2 disease; possibly due to loss of normal NPC2 protein expression in alveolar macrophages and accumulation of functionally inactive cholesterol-rich surfactant.33,34,36 Bronchoalveolar lavage has been reported to improve pulmonary symptoms in children with foam cell infiltrates.37 Histologic features are similar to those seen in patients with NPA and NPB.
GAUCHER DISEASE
Gaucher disease (GD), an autosomal recessive disease, is due to inadequate lysosomal enzyme glucosylceramidase activity, with the resulting accumulation of glucosylceramide, the enzyme’s undegraded substrate, as well as other glycolipids.12,15,38–40 The substrate arises primarily from the breakdown of red blood cells and other tissue cells. The undegraded substrate is then taken up by monocytes and macrophages. Central nervous system involvement with GD may be caused by membrane ganglioside turnover; however, neuron death may also play a role.41
GD encompasses three main clinical subtypes, as well as two additional subtypes, each with characteristic features. Type 1, 2, and 3 GD frequently show lung involvement. Type 1 GD is characterized clinically by bone disease, including osteonecrosis, lytic and sclerotic lesions, and osteopenia; anemia; thrombocytopenia; and hepatosplenomegaly; however, affected patients do not typically show central nervous system involvement. Type 2 and type 3 GD are both characterized by central nervous system involvement with disease; however, type 2 GD patients typically present with disease before age 2, exhibit poor psychomotor development, and rapidly deteriorate, with death occurring usually between ages 2 and 4. Patients with Type 3 GD, in contrast, typically present before age 2, but progress slowly, with some patients surviving to adulthood. There are also two additional subtypes, for which lung involvement is not characteristic. Cardiovascular GD shows mitral value and aortic valve calcification, corneal opacities, and supranuclear opthalmoplegia. Perinatal lethal GD shows nonimmune hydrops fetalis or skin abnormalities.
GD may be caused by mutations in Saposin C and acid-β-glucosidase.42 Diagnosis of GD requires the demonstration of inadequate glucosylceramidase activity in peripheral blood leukocytes or other cells. Treatments include enzyme replacement therapy or substrate reduction therapy.43–46
Pulmonary involvement in patients with GD includes pulmonary hypertension, lobar consolidation, and interstitial lung disease.1,12,15,38,47,48 Pulmonary hypertension is a typical finding in GD patients with hepatic disease, perhaps because of the patients’ inability to detoxify gut-derived factors that affect pulmonary endothelium, with resultant pulmonary hypertension. Pulmonary hypertension may also occur in patients with GD without liver disease.47 Some patients with type 1 GD without pulmonary disease may, nonetheless, exhibit rapid fatigability, possibly due to GD-caused circulatory impairment.12,48,49
Histologically, GD cells infiltrate liver, spleen, and bone marrow. Lung involvement is common in pediatric patients and less common in adults.50 Lung involvement may be clinically significant in as many as one-third of patients.51 GD cells most often involve septal capillaries; this is thought to be the cause of GD-associated pulmonary hypertension.51 Infants and children may also demonstrate intra-alveolar involvement. GD cells have also been found to cause substantial septal thickening and patchy involvement in a lymphatic distribution.51 The characteristic Gaucher cell has a “wrinkled paper’’ appearance (Fig. 67-4), highlighted with Periodic acid-Schiff (PAS) staining. Gaucher cells are CD68 negative, in contrast to alveolar macrophages, which show CD68 positivity.52
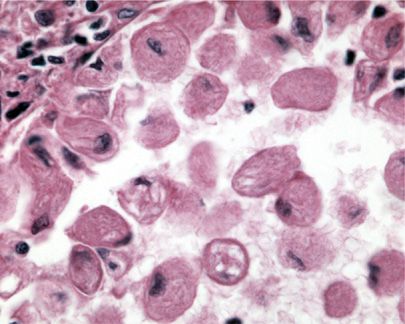
Figure 67-4 High power image of Gaucher cells within an alveolar space showing its characteristic “wrinkled paper’’ appearance.
FABRY DISEASE
Fabry disease (FD) is an X-linked lysosomal storage disease caused by a deficiency of α-galactosidase A enzyme activity, resulting in globotriaosylceramide accumulation in a variety of tissues, including cardiomyocytes, smooth muscle cells, and endothelial cells.53–55
Males with less than 1% enzyme activity develop the classic form of FD, which typically present in childhood or adolescence and is characterized by proteinuria, corneal and lenticular opacities, angiokeratomas, hypohidrosis, and periodic episodes of markedly painful extremities. By the third to fifth decades, gradually worsening renal function usually leads to end-stage renal disease. If an adult with FD survive their renal disease, they typically succumb to cerebrovascular or cardiovascular disorders. If men exhibit greater than 1% enzyme activity, however, they typically exhibit a renal variant, with resultant end-stage renal disease, but without extremity pain or skin lesions. Alternatively, they may exhibit a cardiac variant, which typically presents in men in their 50s to 70s. Findings include proteinuria, mitral value insufficiency, cardiomyopathy, and left ventricular hypertrophy; end-stage renal disease is absent.
Female patients with FD, who are heterozygous for the disease, often exhibit later onset of findings and milder symptoms than males; however, their presentations may range from essentially asymptomatic throughout life to symptoms mimicking their male counterparts with classic FD.
Diagnosis in males is made by showing α-galactosidase A enzyme deficiency in leukocytes, cultured cells, or plasma. Enzyme measurement is not a reliable test for females; some carriers have decreased enzyme activity, but others show normal activity. Enzyme testing for a mutation in GLA, the gene associated with FD, is necessary for diagnosis in female carriers. Males essentially always show a GLA mutation. Enzyme replacement therapy is typically used in male patients and symptomatic female carriers.49,53,54
Pulmonary involvement may occur in both males and females with FD.12,56–58 Patients typically exhibit dyspnea, wheezing, and findings of chronic bronchitis; pulmonary function tests may show obstruction.59 Radiologic changes may be minimal and do not correlate with the severity of pulmonary dysfunction. Histologic diagnosis of FD typically involves identification of laminated inclusions in type II pneumocytes and capillary endothelium.58 Lamellar inclusions have been reportedly identified cytologically in alveolar macrophages, ciliated epithelial cells, and goblet cells by examination of induced sputum, bronchial brushings, and bronchoalveolar lavage specimens.58,60,61
HERMANSKY–PUDLAK SYNDROME
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


