Effects of Neuromuscular Diseases on Ventilation
INTRODUCTION
Neuromuscular diseases comprise a diverse group of disorders that vary markedly in etiology, rate of progression, pattern of respiratory involvement, prognosis, and therapy. Neuromuscular disorders impair the respiratory system as a vital pump; however, depending on the particular disease, the respiratory pump may be impaired at the level of the central nervous system (e.g., cerebral cortex or brainstem), spinal cord, peripheral nerve, neuromuscular junction, or respiratory muscle (Table 84-1).
TABLE 84-1 Levels of Respiratory System Dysfunction Induced by Neuromuscular Diseases and Conditions
The pattern of ventilatory impairment among these disorders is highly dependent on the specific neuromuscular disease. For example, some disorders may impair ventilation at only one level (e.g., isolated diaphragm paralysis) or simultaneously affect it at different levels (e.g., multiple sclerosis [MS]). In addition, the severity of impairment may be minimal and totally resolve with time and proper treatment (e.g., Guillain–Barré syndrome [GBS]) or is characterized by relentless progression to eventual respiratory death (e.g., amyotrophic lateral sclerosis [ALS]). Moreover, some neuromuscular diseases concomitantly affect several structures (e.g., swallowing dysfunction in poliomyelitis, interstitial lung disease in polymyositis), increasing ventilatory workload in patients who already have diminished ventilatory reserve.
This chapter describes the etiology, pathophysiology, and treatment of ventilatory dysfunction in neuromuscular diseases.
RESPIRATORY PATHOPHYSIOLOGY
Substantial information exists concerning the ventilatory function of patients with neuromuscular disease at rest and during sleep, as well as the effects on maximum static inspiratory and expiratory efforts and responses associated with these disorders to hypoxic and hypercapnic challenges. In general, the response of the respiratory system to moderate or severe neuromuscular disease is relatively stereotyped. The typical features are a reduced forced vital capacity (VC), reduced respiratory muscle strength, and, in some cases, malfunction of the neurons that control breathing.
 CONTROL OF BREATHING
CONTROL OF BREATHING
The breathing pattern is often abnormal in patients with neuromuscular disease. In comparison with healthy subjects, patients with respiratory muscle weakness have a low tidal volume and a high respiratory rate that persists in response even to hypoxic or hypercapnic challenge. Moreover, this rapid, shallow breathing pattern is not due to abnormalities in gas exchange (i.e., hypoxemia or hypercapnia) but is more likely to be due to severe muscle weakness and/or disordered afferent and efferent output in motor neurons impaired by the underlying neuromuscular disease.1
Changes in ventilation can be used to evaluate ventilatory drive in subjects with normal lung and respiratory muscle mechanics. However, ventilation is not a good index of respiratory motor activity in subjects with significant respiratory muscle weakness because the thoracic bellows cannot perform increased work of breathing. Decreased ventilatory response to hypoxic or hypercapnic challenge in these patients could indicate abnormalities in afferent information from diseased respiratory muscles, abnormal lung or chest wall mechanics, or upper motor neuron dysfunction, rather than an abnormality in the central control of breathing. In some neuromuscular diseases, degenerative changes in the muscle spindle, impaired afferent stimulation from abnormal stretch reflexes in the muscle spindles, or decreased mechanoreceptor output from tendons may explain the altered breathing pattern.1
Measurement of mouth occlusion pressure generated during the first 100 ms of inspiration (P0.1) is relatively independent of inspiratory effort and therefore is a more reliable estimate of central ventilatory drive independent of respiratory muscle mechanics.2 P0.1 is maintained or increased in patients with neuromuscular disease despite substantial muscle weakness. The relationship between respiratory mechanics, respiratory muscle strength, and control of ventilation has been examined in patients with neuromuscular diseases in comparison with healthy control subjects. Although patients had 37% and 52% reductions in maximum inspiratory and expiratory mouth pressures, respectively, their P0.1 was 66% greater than that of controls.1
Similar findings were encountered when normal subjects had acute muscle weakness induced by curarization. After severe muscle weakness was induced, significant increases in P0.1 were observed during hypercapnic challenge.3 Partial curarization of spontaneously breathing cats also produced a marked increase in phrenic nerve discharge despite a substantial decrease in minute ventilation.4 These studies indicate that under conditions of substantial respiratory muscle weakness, ventilation is not a reliable measure of central respiratory drive, and that central respiratory drive, at least when measured by P0.1, is usually well preserved.
 RESPIRATORY MUSCLE FUNCTION
RESPIRATORY MUSCLE FUNCTION
Patients with neuromuscular disease who develop significant respiratory muscle weakness may experience fatigue, dyspnea and impaired control of secretions, recurrent lower respiratory tract infections, acute or chronic presentations of respiratory failure, pulmonary hypertension, and cor pulmonale.
The pattern, prognosis, and degree of respiratory muscle weakness attributable to a neuromuscular disorder are varied. They depend on the level of neuromuscular system impairment, the prognosis of the underlying disorder, and whether therapy is available. Patients with neuropathy, such as GBS, tend to have less severe respiratory muscle weakness than patients with lower motor neuron lesions or neuromuscular junction disorders, such as myasthenia gravis. Even when respiratory muscle dysfunction is observed, not all respiratory muscles are equally impaired, and the course of the underlying neuromuscular disease and degree of respiratory and nonrespiratory muscle impairment can be very different between patients with the same disease. In some neuromuscular disorders, respiratory muscle weakness is the only presentation of an underlying disease (i.e., neuralgia amyotrophy of the diaphragm); in the case of muscular dystrophy, significant respiratory muscle weakness may occur only late in the disease course. Severe, relentless, progressive dysfunction of the respiratory muscles may occur, as in ALS, or be characterized by exacerbations and relapses (e.g., MS). Finally, respiratory muscle weakness may completely reverse with time (phrenic nerve injury after open heart surgery) or with therapy (plasmapheresis in myasthenia gravis).
A significant proportion of patients with severe respiratory muscle weakness were also found in 50% of 30 asymptomatic patients with stable chronic neuromuscular disease. Reductions in inspiratory and expiratory mouth pressures did not correlate with general muscle strength assessment; however, the type of neuromuscular disease and distribution of general muscle weakness both correlated with respiratory muscle impairment.5 Patients with myopathy, rather than polyneuropathy, whose involvement produced proximal rather than distal limb muscle weakness, were more likely to have significant respiratory muscle weakness. Pulmonary symptoms correlated poorly with evidence of respiratory muscle weakness.
Explanations for the lack of pulmonary complaints in these two studies despite significant muscle weakness are not clear. Patients with chronic and severe neuromuscular disease are usually sedentary and incapable of exertion and, therefore, seldom stress the respiratory system, which may explain their lack of symptoms.
The rapid, shallow breathing pattern found in patients with respiratory muscle weakness may be due to decreased respiratory muscle force generation, but it may also be due to changes in lung and chest wall elastic recoil. A decrease in inspiratory muscle tone may lead to unopposed lung elastic recoil, which reduces lung volume and produces chronic changes in chest wall tone and distensibility. Once inspiratory muscle strength decreases to approximately 30% of normal, abnormalities in gas exchange (manifested primarily by hypercapnia) commonly occur.
Expiratory muscle weakness is also commonly observed in patients with neuromuscular disease. It causes ineffectual cough and impaired secretion clearance, which in some patients lead to recurrent lower respiratory tract infections. In normal persons, dynamic compression of the central intrathoracic airways by large changes in pleural pressure generated by forceful contraction of the expiratory muscles acts to propel secretions proximally, where they can be expectorated. As expiratory muscle weakness progresses, pleural pressures generated during coughing efforts are reduced and airway clearance is impaired.
 LUNG AND CHEST WALL MECHANICS
LUNG AND CHEST WALL MECHANICS
A characteristic hallmark of chronic neuromuscular disease is a decreased VC. The VC is reduced because of respiratory muscle weakness, and the decrease in VC parallels the progression of the underlying disease, but the magnitude of the reduction in VC is greater than expected solely based on the reduction in respiratory muscle force. The sigmoidal shape of the pressure–volume curve would suggest that large reductions in pressure initially produce only small reductions in lung volume. In 25 patients with a variety of neuromuscular diseases, De Troyer et al.6 found that reductions in VC were much greater than expected, solely based on the reductions in inspiratory muscle strength (Fig. 84-1).
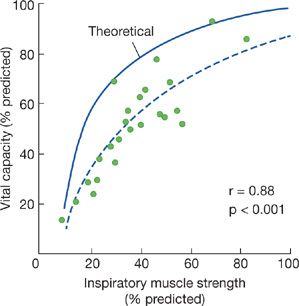
Figure 84-1 The solid curve represents the theoretic effect of respiratory muscle weakness on vital capacity (VC) on the assumption that the relaxation pressure–volume characteristic of the lung and chest wall are normal and that the inspiratory and expiratory muscles are uniformly involved. Dashed curve is the logarithmic regression calculated in 25 patients with neuromuscular disease (closed circles). Data suggest that loss of lung volume is out of proportion to the degree of inspiratory muscle weakness. (Data from De Troyer A, Borenstein S, Cordier R. Analysis of lung volume restriction in patients with respiratory muscle weakness. Thorax. 1980;35:603–610.)
It appears that in addition to muscle weakness, alterations in the mechanical properties of the lung and chest wall contribute to the reduced VC. Using the mean deflationary pressure–volume curve of the lung in 25 patients with moderate-to-severe neuromuscular disease, De Troyer et al.6 found, on average, a 40% decrease in lung compliance (Fig. 84-2). Furthermore, measurements of static lung compliance measured during inspiration in patients with neuromuscular diseases also show marked reductions, suggesting that chronic respiratory muscle weakness changes the elastic properties of the lung itself.7
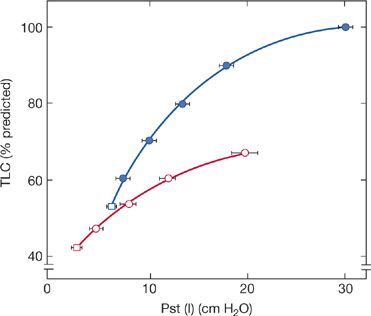
Figure 84-2 Static expiratory pressure–volume curve in patients with neuromuscular disease and respiratory muscle weakness. Open circles represent average data in 25 patients. Volume is displayed on the Y-axis as a percentage of predicted total lung capacity (TLC). Closed circles represent mean predicted values. In patients, absolute lung volume was decreased for any given transpulmonary pressure. (Data from De Troyer A, Borenstein S, Cordier R. Analysis of lung volume restriction in patients with respiratory muscle weakness. Thorax. 1980;35:603–610.)
The cause of reduced lung distensibility in patients with neuromuscular disease is unknown. Several causes such as failed maturation of normal lung tissue during childhood or congenital neuromuscular diseases, the presence of micro- or macroatelectasis, increased alveolar surface tension caused by breathing chronically at low tidal volumes, and alteration in lung tissue elasticity have been proposed.
Impaired lung maturation is unlikely, since patients who develop neuromuscular disease in adulthood also have a reduction in VC that is disproportionate to the magnitude of respiratory muscle weakness. The presence of micro- and macroatelectasis also appears untenable, because most patients who have significant reductions in VC typically do not have alveolar collapse on radiographic imaging. Although rapid and shallow breathing patterns are encountered in patients with chronic severe neuromuscular disease, mechanical hyperinflation of the lung does not restore lung distensibility. Therefore, increased alveolar surface tension is not considered the principal cause of reduced lung compliance in patients with chronic neuromuscular disease.
Theoretically, a reduction in lung tissue elasticity may also contribute to a reduction in lung compliance in patients with neuromuscular disease, but there is no evidence that lung collagen, elastin, and other matrix proteins change in these diseases. Currently, the reason for the reduction in lung compliance in patients with chronic neuromuscular disease is unknown and awaits further study.
Many studies indicate that chest wall compliance is decreased by approximately 30% in patients with chronic neuromuscular disorder. In 16 patients with chronic neuromuscular diseases (e.g., spinal cord injury, Duchenne muscular dystrophy [DMD], and myasthenia gravis), the weighted spirometer technique was used to examine chest wall compliance in comparison with that of 20 healthy control subjects. The weighted spirometer technique delivers an airway pressure that causes an increment in thoracic volumes so as to construct the pressure–volume relationship. In 12 of these patients, chest wall compliance was reduced (Fig. 84-3).8 Based on the contour of the pressure–volume curve of the normal relaxed chest wall at lower lung volumes, a reduction in functional residual capacity (FRC), as seen in patients with chronic neuromuscular diseases, may in itself reduce static chest wall compliance. However, in other disorders in which FRC is decreased owing to parenchymal lung disease (i.e., pulmonary fibrosis), a reduction in chest wall compliance has not been demonstrated. The mechanism for the reduction in chest wall compliance in patients with chronic neuromuscular disease has not been definitely established, but limitations in respiratory excursions have been proposed to lead to increased rib cage stiffness by decreasing the viscoelasticity of chest wall structures. Regardless of the mechanism, it appears that a reduction in chest wall compliance, along with a decrease in lung compliance, contributes to the marked decrease in VC observation in patients with neuromuscular disease.
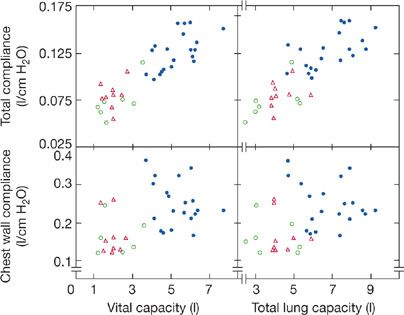
Figure 84-3 Relationships between total respiratory system compliance and VC and TLC (upper panels) and between chest wall compliance and VC and TLC (lower panels) in 16 patients with chronic neuromuscular diseases (open symbols) compared to 20 healthy controls (closed circles). Triangles symbolize patients who are quadriplegic, squares symbolize patients who are paraplegic and four patients had generalized neuromuscular diseases (circles). Compared to healthy controls (closed circles), those with neuromuscular disease had significant reductions in both total respiratory system and chest wall compliance. (Data from Estenne A, Heliporn A, Dellez L, et al. Chest wall stiffness in patients with chronic respiratory muscle weakness. Am Rev Respir Dis. 1983;128:1002–1007.)
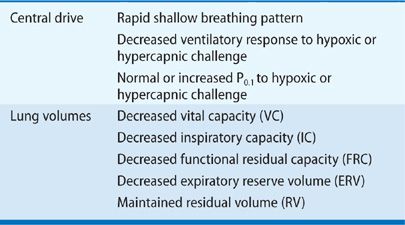
Although reductions in VC appear to be clearly established in patients with chronic neuromuscular disease, the effect of chronic neuromuscular disease on FRC and residual volume (RV) are contradictory. FRC and RV have been reported to be unchanged, decreased, or mildly increased. Discrepancies between these studies could be explained by differences in the type of severity, and stages of neuromuscular diseases studied or body positions in which testing were performed. However, in two separate studies, patients with a wide variety of chronic neuromuscular diseases, all studied in a similar seated position, were found to have approximately 20% reductions in FRC but normally predicted values of RV.9,10 Furthermore, confirmation of these findings was demonstrated in eight patients with myasthenia gravis given pyridostigmine, which acutely decreased FRC by approximately 15% without any significant change in RV.11 On the basis of the previously mentioned data, it appears that patients with chronic neuromuscular disease have moderate reductions in VC and total lung capacity (TLC) that are associated with a moderate decrease in FRC and a normal RV. The decrease in VC not only is due to respiratory muscle weakness but also appears to result from decreased lung and chest wall compliance. Table 84-2 summarizes the effect of neuromuscular diseases on both lung volumes and central respiratory drive.
 SLEEP-RELATED BREATHING DISTURBANCES
SLEEP-RELATED BREATHING DISTURBANCES
Breathing during sleep is often abnormal in patients with neuromuscular disease. Impaired sleep quality and hypopnea and hypercapnia related to rapid eye movement (REM) sleep are frequent. Patients with chronic neuromuscular disease of various causes have significant and numerous episodes of nocturnal desaturation, which are most prevalent during REM sleep and are characterized by hypoventilation rather than upper airway obstruction (Fig. 84-4).12 Of six patients, 16 to 22 years of age, with advanced DMD, randomized to breathing either air or oxygen on two consecutive nights, five demonstrated significant oxygen desaturation during REM sleep and approximately 35% reductions in minute ventilation compared to their baseline awake values. Furthermore, the severity of diaphragmatic dysfunction was related to the degree of oxygen desaturation.13
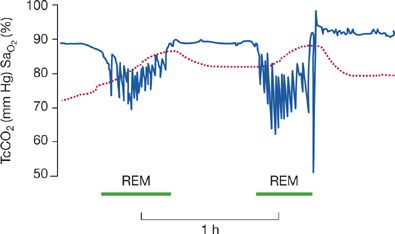
Figure 84-4 Oxygen desaturation and hypercapnia in REM sleep shown from a recording of an all-night sleep study. Transcutaneous carbon dioxide (TcCO2) is shown as the dashed line; arterial hemoglobin oxygen saturation (SaO2) is shown in the line with sharp deflections. Patients with neuromuscular diseases are often at risk for hypoventilation during REM sleep resulting in a decrease in SaO2 and increase in TcO2. (Data from Bye PT, Ellis ER, Issa FG, et al. Respiratory failure and sleep in neuromuscular disease. Thorax. 1990;45:241–247.)
Several hypotheses have been proposed to explain nocturnal desaturation. Patients with chronic neuromuscular diseases develop an even more rapid and shallow breathing pattern during REM sleep. A rapid and shallow breathing pattern leads to increased dead space ventilation, which promotes hypercapnia and worsened oxygenation. Reductions in ventilatory drive may be accentuated during sleep in patients with underlying neuromuscular disease, especially in those who have pre-existing abnormalities of ventilatory control, which may further contribute to worsened nocturnal hypoventilation.
It has been hypothesized that patients with neuromuscular disease, especially with diaphragmatic dysfunction, may be more prone to nocturnal desaturation during REM sleep. Intercostal muscle and accessory respiratory muscle activity during REM sleep are depressed, with a greater contribution of the diaphragm required for maintenance of eucapnia and oxygenation. Support for this hypothesis comes from studies that have found diaphragm dysfunction to be highly correlated with the presence and magnitude of REM-related oxygen desaturation. A direct relation has been found between the lowest SaO2 value measured during REM sleep and the percentage fall in VC measured between the erect and supine positions, using the latter measurements as an index of diaphragm weakness. Similarly, among patients who have paradoxical abdominal movement, signifying a decrease in diaphragmatic contribution to ventilation, a greater oxygen desaturation in both REM and non-REM sleep is observed.14 In contrast, patients with isolated diaphragmatic dysfunction with intact accessory muscle function are not predisposed to severe nocturnal hypoventilation.15 Accordingly, severe hypoventilation may become evident only when diaphragmatic weakness is found in the background of global accessory and intercostal muscle weakness, or when ventilatory reserve is severely reduced for other reasons, such as asthma or chronic obstructive pulmonary disease (COPD).
Abnormalities in nocturnal gas exchange are harbingers of problems in daytime gas exchange. Hypoventilation during sleep precedes the appearance of daytime hypercapnia, and patients with the most impaired gas exchange during REM sleep have the greatest degree of daytime hypercapnia. Moreover, patients with normal nocturnal gas exchange are unlikely to have abnormal daytime values. Noninvasive (e.g., nasal positive-pressure ventilation, external negative-pressure ventilation) or invasive (e.g., positive-pressure ventilation by tracheostomy) mechanical ventilation improves nocturnal gas exchange and sleep quality, with simultaneous improvement in daytime gas exchange.
Two theories have been proposed to explain the sustained improvements in gas exchange during daytime spontaneous breathing in patients with chronic neuromuscular disease who receive nocturnal ventilatory support. One theory states that nocturnal ventilation rests chronically fatigued respiratory muscles, thereby permitting improved spontaneous ventilation and gas exchange. Although several studies have demonstrated that noninvasive ventilation provides inspiratory muscle fatigue in patients with neuromuscular disease, or that mechanical ventilation consistently increases respiratory muscle strength. However, evidence that inspiratory muscle fatigue is commonly present in patients with neuromuscular disease or that mechanical ventilation consistently increases respiratory muscle strength is lacking. An alternative hypothesis suggests that nocturnal ventilatory support lowers the central respiratory center CO2 set point and, thereby, sets the central controller to maintain a lower spontaneous daytime CO2 level. This hypothesis is supported by studies showing that after several weeks of chronic nocturnal ventilation, hypoventilation was less severe in nocturnal studies without ventilation than it had been on baseline nights before chronic intermittent ventilation. Moreover, interruption of successful nocturnal noninvasive ventilation in patients with neuromuscular disease and chronic respiratory failure results in a return of nocturnal hypoventilation and symptoms of impaired gas exchange without evidence of respiratory muscle dysfunction. To date, neither of the previously mentioned theories has been established conclusively, and further investigation is warranted, as one or the other, or both, may be valid in different patients.
ASSESSMENT OF RESPIRATORY FUNCTION
Patients with significant respiratory muscle impairment may range from being totally asymptomatic to having moderate dyspnea at rest or, in some cases, overt respiratory failure. Some patients with neuromuscular disease may have significant weakness of the respiratory muscles and be asymptomatic,16 whereas others may present with ventilatory failure without an established history of a neuromuscular disease. In the latter patients, the diagnosis of neuromuscular disease may initially be entertained only after difficulty is encountered in weaning the patient from mechanical ventilation. A detailed history and physical examination, coupled with appropriate diagnostic tests, enable the physician to diagnose the presence and type of neuromuscular disease and its effect on the respiratory system. The following section reviews features of the history and physical examination and the diagnostic studies considered useful in the assessment of respiratory function in patients with neuromuscular disease.
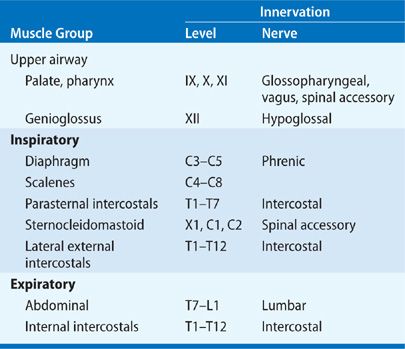
In order to provide an organized approach to direct the clinical history taking and physical examination of patients with neuromuscular disease, Table 84-1 characterizes the types of neuromuscular diseases that present at different levels of the neuromuscular system, and Table 84-3 describes the innervation of the different groups of respiratory muscles.
 CLINICAL HISTORY
CLINICAL HISTORY
The signs and symptoms of respiratory muscle weakness due to a neuromuscular disease are usually nonspecific and of limited value. Moreover, the clinical manifestations of respiratory muscle dysfunction depend on the specific muscle or muscles affected and the extent of their impairment. In conditions of mild weakness, or in the early stages of neuromuscular disease, the patient may be totally asymptomatic.16 As respiratory muscle weakness progresses, however, dyspnea on exertion, followed by dyspnea at rest occurs. Disturbances in sleep and daytime hypersomnolence resulting from nocturnal hypoventilation may occur, and if the expiratory muscles are affected, patients may have impaired cough and repeated lower respiratory tract infections. As respiratory muscle weakness becomes more severe, hypercapnia or hypoxemia becomes evident and respiratory failure may ensue, requiring ventilatory support.
The clinical history is invaluable in that it may be the first clue that a neuromuscular disease is the cause of the patient’s pulmonary dysfunction. A history is also useful in characterizing the type of neuromuscular disease that is present. Dyspnea and impaired cough with or without recurrent lower respiratory tract infections may be the first clinical clues that a neuromuscular disease is present. Impaired swallowing due to bulbar symptoms and the presence of peripheral limb muscle weakness are indications that one is dealing with generalized neuromuscular disease.
 PHYSICAL EXAMINATION
PHYSICAL EXAMINATION
Although the physical examination may yield normal results in patients with early or mild impairment of the respiratory system, patients with more established disease often demonstrate tachypnea at rest. Further clinical information on the nature of the underlying disease and the extent of underlying muscle impairment can be gleaned from the pattern of respiratory muscle contraction in both seated and supine positions. Respiratory rate should be recorded along with any evidence of nasal flaring, intercostal muscle retraction, or palpable evidence of contraction of the sternocleidomastoid and scalene muscles. Furthermore, inward paradoxical motion of the rib cage or abdomen should be sought, as its presence may indicate a respiratory workload that is greater than the patient’s respiratory muscle strength, or evidence of severe weakness of the diaphragm as a result of the underlying neuromuscular disease. Besides gross paradoxical movement of the rib cage or abdominal compartments, asynchronous compartmental movements (e.g., one compartment moving faster than the other) may be early evidence of impaired respiratory pump performance.
The hallmark finding of severe diaphragm weakness or paralysis is paradoxical inward movement of the abdomen with inspiration. In the presence of severe diaphragm weakness, the upper abdomen moves inward when the upper rib cage moves outward in stark contrast to the normal pattern of synchronized outward movements of the rib cage and abdominal compartments. Besides paradoxical movement of the upper abdomen, a marked increase in respiratory rate, accompanied by progressive accessory muscle use, and increased dyspnea occur when patients assume the recumbent position due to hypoxemia, hypercapnia, and placing the accessory inspiratory muscles at mechanical disadvantage. Upon reassuming the upright posture, patients may have palpable phasic contractions of the abdominal expiratory muscles. Physiologically, this inward movement of the abdomen on expiration enables passive outward movement of the upper abdomen and diaphragm descent during expiratory muscle relaxation in early inspiration.
Besides a detailed examination of the respiratory musculature and breathing pattern, the physical examination should include a complete neuromuscular examination to exclude systemic involvement. Inspection for atrophy or fasciculations of respiratory and nonrespiratory muscles may point to a lower motor neuron disease. The presence of scoliosis may contribute to the development of restrictive ventilatory pattern.
 RADIOGRAPHIC ASSESSMENT
RADIOGRAPHIC ASSESSMENT
In patients with severe inspiratory muscle weakness or bilateral diaphragm paralysis, maximum inspiration is limited and lung volume appears reduced on chest radiograph. Unilateral hemidiaphragm paralysis produces an elevated hemidiaphragm on the affected side.17
Fluoroscopy is often used in the assessment of diaphragm paralysis with the patient making a forceful sniff in the supine position.17 In unilateral diaphragm paralysis, a positive “sniff” test may demonstrate paradoxical upward movement of the affected hemidiaphragm. However, “sniff” tests have a false-positive rate as high as 6% in normal persons. The use of the “sniff” test to diagnose bilateral diaphragm paralysis is limited by compensatory abdominal muscle contraction. With abrupt cessation of abdominal muscle contraction during early inspiration, the abdominal contents descend caudally. The abdominal wall moves outward and the diaphragm will then appear to descend caudally, at least radiographically. Besides the fact that passive diaphragm descent due to active abdominal muscle contraction is a limitation during fluoroscopy, the fluoroscopic observational field used to examine the diaphragm is limited because of the small visual band that encompasses only the diaphragmatic dome and adjacent ribs. If rib cage rostral movement exceeds diaphragm ascent, the diaphragm will appear to descend lower than the thorax that may falsely suggest the presence of diaphragm shortening.18
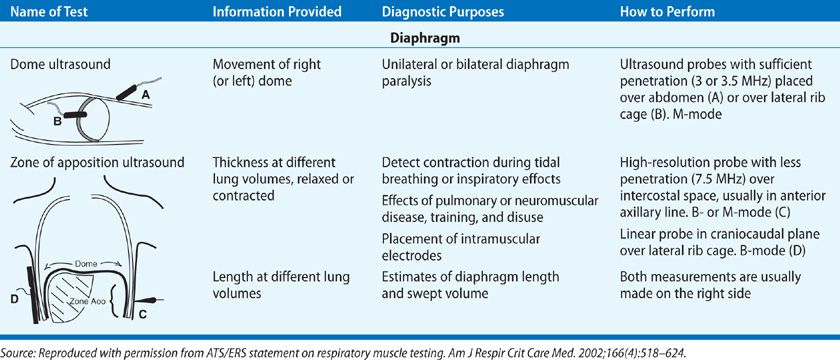
Although the diaphragm itself is poorly echogenic, ultrasound can be used to assess its function because the parietal pleura and peritoneal membranes lining the diaphragm are brightly echogenic. The two approaches used are the visualization of the dome or measurement of the muscle thickness at the zone of apposition.2 Craniocaudal movement of the dome of the diaphragm can be measured by placing an ultrasound probe on the upper abdomen or on the lateral chest as shown in Table 84-4. This technique has compared favorably to the traditional fluoroscopic procedures used to assess diaphragm movement. Because the costal portion of the diaphragm is close to the skin, the zone of apposition (Table 84-4) is an ideal area to use ultrasound for assessment of the diaphragm thickness and estimation of length. The thickness of the diaphragm increases with increasing lung volumes and is inversely proportional to its length. Measurement of the zone of apposition will permit the detection of diaphragm contraction during inspiratory efforts when trying to diagnose diaphragm paralysis. As the subject with diaphragm paralysis makes an inspiratory effort there will not be thickening of the diaphragm at the zone of apposition. Measurement of the thickness also allows for the assessment of atrophy or the effect of neuromuscular diseases.
TABLE 84-4 Two Approaches to Ultrasound of Diaphragm. Taken from the American Thoracic Society Consensus Statement on Respiratory Muscle Testing
 ARTERIAL BLOOD GAS ANALYSIS
ARTERIAL BLOOD GAS ANALYSIS
Arterial blood gas abnormalities usually occur only in patients with severe respiratory muscle weakness. Hypoxemia is usually mild and may occur as a result of macroatelectasis and subsequent intrapulmonary shunting or ventilation–perfusion mismatch. In addition, patients with impaired muscle strength have impaired cough and may retain secretions that further contribute to the development of hypoxemia. Measurement of arterial oxyhemoglobin saturation by pulse oximetry, which has become an extremely common laboratory test for oxygenation, is an insensitive indicator of hypoventilation. In patients with mild-to-moderate respiratory muscle weakness, the value of solely measuring the level of oxygenation is limited and may be misleading.
Hypercarbia is an insensitive measure of respiratory muscle strength. The PaCO2 does not increase until respiratory muscle strength (measured by maximum inspiratory and expiratory mouth pressures) is less than 50% of predicted. In patients with severe respiratory muscle weakness, however, an elevation in PaCO2 may be evident. Serum bicarbonate and arterial pH values may help determine whether an acute or chronic respiratory acidosis is present. Because daytime hypercapnia is usually followed by nocturnal hypoventilation, the presence of daytime hypercapnia should prompt investigation of the breathing pattern and gas exchange during sleep, so that appropriate therapy (e.g., nocturnal supplemental oxygen or noninvasive ventilation) can be implemented.
RESPIRATORY MUSCLE STRENGTH
A variety of techniques have been used to assess respiratory muscle strength. Each is discussed below.
 MAXIMUM MOUTH PRESSURES
MAXIMUM MOUTH PRESSURES
Maximum static inspiratory and expiratory mouth pressures, measured at the airway opening during a voluntary contraction against an occluded airway, are the simplest and most commonly performed tests of respiratory muscle strength. Although several methods exist, the technique of Black and Hyatt is still the most widely used.19 In this technique, mouth pressures are measured using a handheld manometer with the patient seated upright with a nose clip on. During these maneuvers, the patient purses the lips inside a circular wide-bore rubber mouthpiece, which prevents perioral air leakage. This small orifice (2 mm in diameter, 15 mm in length) is placed in the circuit to minimize the contribution of the facial muscles to airway pressure and to keep the glottis open. Maximum inspiratory pressures (PImax) are measured near RV after maximal expiration, while maximal expiratory pressures (PEmax) are measured at or near TLC. In each case, efforts are maintained for at least 1 second. Maximum inspiratory and expiratory mouth pressures in normal males and females are listed in Table 84-5. Reported values in normal subjects vary widely and may be due to differences in techniques between different studies or a learning effect in subjects who perform these maneuvers.20–25
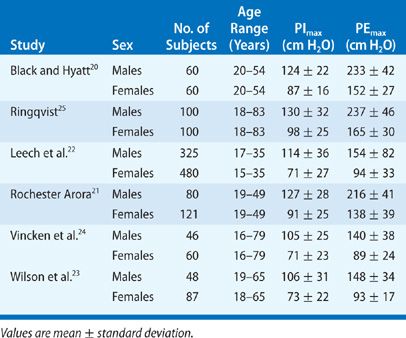
A major factor affecting PImax is lung volume. PImax is greatest at RV, whereby the inspiratory muscles are at greatest mechanical advantage and the outward elastic recoil of the respiratory system is maximum. On the other hand, measurement of PEmax is greatest at TLC because expiratory muscles are at greatest mechanical advantage and inward elastic recoil of the respiratory system is greatest (Fig. 84-5). Only at FRC, where the respiratory system recoil pressures measured at the airway opening are zero, are maximum inspiratory and expiratory mouth pressures solely a function of the pressure generated by actively contracting respiratory muscles (Pmus).
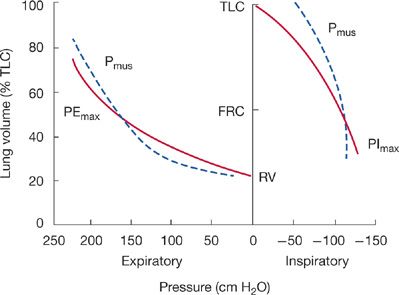
Figure 84-5 The effect of lung volume on maximum respiratory pressures (PImax and PEmax) measured at the airway opening displayed by solid lines. Both PImax and PEmax are made up of two components: the pressure generated by the respiratory muscles (Pmus, dashed lines) and the recoil pressure of the respiratory system. Only at FRC when recoil pressures are zero are PEmax and PImax solely due to Pmus.
Changes in lung volume due to chest wall or lung pathology may have important effects on the generation of maximum respiratory pressures in patients. For example, patients with COPD and significant hyperinflation have a larger FRC and RV than normal subjects; therefore, PImax performed at FRC or RV usually results in lower values than in age- and sex-matched normal subjects. Likewise, a reduction in TLC due to restrictive ventilatory diseases may result in a reduction in measured values for PEmax. Therefore, it is important to realize that in patients with pathologically altered lung volumes, all or part of the reduction in mouth pressures may be due to inspiratory muscle mechanical disadvantage.
Maximum inspiratory and expiratory mouth pressures in patients with neuromuscular diseases range from normal to severely reduced. Patients may have significant respiratory muscle weakness without any pulmonary complaints, and no correlation exists between respiratory muscle strength and the presence of generalized nonrespiratory muscle weakness.16 When PImax falls below 30 cm H2O, ventilatory failure commonly ensues.
The assessment of a patient’s ability to generate an effective cough is extremely important when managing the pulmonary effects of neuromuscular diseases. Nearly all of these disorders result in weak cough, which puts the individual at risk for aspiration and pneumonia. While a normal PEmax ensures that the patient has adequate cough, a low PEmax could result from poor effort, bulbar weakness not allowing a tight seal around the mouthpiece or true expiratory muscle weakness. Therefore there is interest in developing a test that will allow the assessment of cough strength in a nonvolitional manner. Measurement of positive pleural pressures with an esophageal balloon during a forceful cough (Pes cough) has also been proposed as a measure of expiratory muscle strength. Pes cough has been shown to decrease in parallel with PEmax when expiratory muscle weakness is induced by progressive curarization. A study examined the use of measurement of gastric pressures during cough (Pga cough) in a group of normal subjects and in those with suspected respiratory muscle weakness from pulmonary and neuromuscular disease. The measurement of Pga cough is theoretically better because it takes into account the abdominal musculature, eliminates the problem of leak around the mouthpiece, and a cough maneuver is easier to perform than the PEmax maneuver. In 122 patients with a normal PEmax, more than 95% also had a normal Pga cough, but in 171 patients with a low PEmax 72 had a normal Pga cough suggesting a high false-positive rate of a low PEmax. Conversely, in 105 patients with a low Pga cough only 6 had a normal PEmax suggesting a low false-positive rate for a low Pga cough.31
Transdiaphragmatic Pressure Measurement
While maximum static airway pressures are useful measures of global respiratory muscle strength, they fail to assess individual respiratory muscle function. Since the diaphragm is the primary muscle of inspiration, and may be susceptible to isolated disease (e.g., phrenic nerve paralysis after open heart surgery or idiopathic diaphragm paralysis), specific testing of diaphragm strength is desirable in some patients. Assessment of diaphragm strength is made by measuring gastric (Pga) and endoesophageal (Pes) pressures with balloon-tipped catheters placed in the stomach and midesophagus, respectively. Transdiaphragmatic pressure (Pdi) is then calculated as the algebraic subtraction of Pes from Pga (Pdi = Pga – Pes).2
Maneuvers to elicit maximum transdiaphragmatic pressures (Pdi,max) have been the subject of intensive study. Earlier studies measured Pdi during maximum static inspiratory efforts against a closed airway (e.g., Muller maneuver) at FRC or RV. However, this maneuver results in submaximal diaphragm activation, with the degree of activation varying widely from subject to subject. Several studies have demonstrated significant intraindividual variability, with a coefficient of variation as high as 40% in measurement of Pdi,max during Muller maneuver. When five maneuvers to measure Pdi,max in 35 subjects (10 normal, 13 with restrictive lung disease, and 12 with COPD) were compared, a combined maneuver of active expulsion with superimposed Muller maneuver yielded the most reproducible and maximal transdiaphragmatic pressure.32
Phrenic Nerve Stimulation
A crucial factor in the measurement of diaphragm strength is the ability to consistently obtain maximal activation of the diaphragm during volitional efforts. Electrophrenic stimulation is a method that has been utilized to consistently activate the diaphragm. Besides assessing diaphragm strength, this technique has the added advantage of assessing phrenic nerve conduction and excluding the possibility of phrenic nerve injury in patients with diaphragm weakness of unknown origin.2
The phrenic nerve is stimulated in the neck near the posterior border of the sternocleidomastoid muscle, at the level of the cricoid cartilage, where the phrenic nerves are most superficial. Stimulation may be performed either transcutaneously with surface electrodes (electrical stimulation electrodes), magnetic coil, or percutaneously with needle or wire electrodes. The percutaneous method is rarely used now. Stimulation of the phrenic nerves must be supramaximal with regard to voltage and current. Supramaximal conditions are ensured by increasing the stimulus intensity until maximum diaphragm muscle action potential (DMAP) or Pdi is achieved. The DMAP is measured by surface electromyography (EMG) electrodes, and the Pdi is measured by measuring the esophageal and gastric pressures via two pressure transducers as described earlier. The DMAP is then checked periodically throughout the study to ensure that consistent stimulation is maintained.2,33
The most commonly used technique of electrophrenic stimulation now employs a frequency of one pulse per second to measure Pdi during a single unfused twitch contraction (e.g., Pdi,tw). Pdi,tw has also been used to assess maximal static Pdi indirectly by the twitch occlusion technique.2,34 In this method, single twitches are superimposed on progressively stronger voluntary Pdi contractions. As voluntary effort and Pdi increase, the increment in Pdi produced during the twitch (the twitch deflection superimposed on the Pdi) decreases (Fig. 84-6A). When there is no discernible Pdi,tw deflection, it is assumed that the diaphragm is maximally activated and voluntary efforts from the inverse linear relationship between the amplitude of the superimposed twitch and Pdi measured during volitional effort. The extrapolation of the line of this relationship to the X-axis has been interpreted as representing maximum static Pdi (Fig. 84-6B).
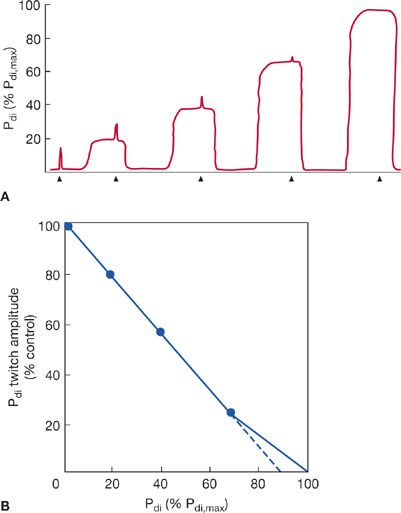
Figure 84-6 A. Illustration of a typical Pdi tracing during twitch occlusion study. As the Pdi increases during volitional efforts, the superimposed Pdi deflection during phrenic nerve 1-Hz stimulation (twitch) decreases. At 100% of Pdi,max, the diaphragm is maximally activated and no superimposed twitch is seen. Arrows on the horizontal axis mark indicate the phrenic nerve twitches. B. Data from A plotted as Pdi,tw amplitude versus voluntary Pdi. Using linear regression, Pdi,max can be extrapolated from results obtained during submaximal efforts. It has been suggested that extrapolation performed from Pdi values below 70% of maximum may underestimate Pdi,max by approximately 10% (dashed line).
An alternate way to perform phrenic nerve stimulation is via magnetic stimulation.2,35 In this technique, an electric current is run through a coil thereby producing a magnetic field. The coil is placed over the spinous process of the seventh cervical vertebral body (cervical magnetic stimulation) stimulating the C3 to C5 cervical roots of the phrenic nerve causing the diaphragm to contract. Magnetic stimulation of this area will also stimulate contraction of neck and upper rib cage muscles as well. The advantages of this technique are that it is less painful than the electrical stimulation method, and it is easier to evoke diaphragm contractions. Also it is possible to perform magnetic stimulation of the phrenic nerve while the patient is in the supine position by placing the magnetic coil anterior to the sternum. This would allow for hospitalized bed-bound patients to be evaluated for diaphragm weakness via phrenic nerve stimulation. One of the disadvantages of magnetic stimulation is that it lacks the specificity that electrophrenic stimulation has for the diaphragm and obtaining an EMGdi signal can be more difficult with magnetic stimulation. When comparing magnetic stimulation Pdi,tw to electrophrenic Pdi,tw, the Pdi,tw tends to be 20% to 25% higher with magnetic stimulation.
In addition to assessing diaphragm strength, phrenic nerve stimulation can be used to assess phrenic nerve function. The EMGdi is measured via surface or esophageal electrodes during electric or magnetic phrenic nerve stimulation and the phrenic nerve conduction time can be measured.2 This measurement is useful when assessing possible injury to the phrenic nerve from thoracic surgery, trauma, or neuropathies (i.e., critical illness polyneuropathy [CIP] or GBS). The normal conduction time via electrical stimulation is 7.5 to 9 ms, but the normal conduction time via magnetic stimulation has not been well defined because activation of the brachial plexus affects the phrenic nerve conduction time. However, Luo et al. demonstrated that if the magnetic coil is placed anteriorly to the cricoid cartilage then the phrenic nerve conduction time was very similar to that obtained by electric stimulation. The authors believe that this occurred because there was more brachial plexus activation in the lower position compared to the higher position.36
Because of the relative invasiveness of electrophrenic stimulation of the diaphragm, and the large coefficient of variation in some studies when Pdi was measured during maximal volitional efforts, some investigators prefer measuring maximum inspiratory pressures during a sniff maneuver (Fig. 84-7). In this technique, the subject performs a vigorous sniff against an unoccluded airway. During such an effort, the nose acts as a Starling resistor, thereby generating intrathoracic pressures against an occluded airway. Some investigators argue that this maneuver approaches a more natural respiratory effort than other types of maneuvers used to measure maximum inspiratory pressures and thus should be easily mastered by patients and more reproducibly performed by technicians.37
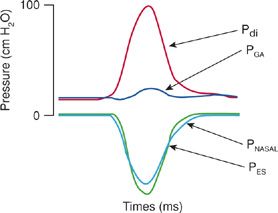
Figure 84-7 Pressure tracing produced utilizing sniff maneuver in a normal individual with gastric and esophageal balloon manometry in place. The noninvasive PNASAL pressure closely mirrors that of the invasive PES. PGA, gastric pressure; Pdi, transdiaphragmatic pressure; PES, esophageal pressure; PNASAL, nasal pressure during sniff maneuver.
Analysis of Rib Cage and Abdominal Motion
During normal tidal breathing, the chest and abdominal compartments move synchronously in an outward direction, owing to diaphragm contraction decreasing pleural pressure and increasing abdominal pressure. In situations where the diaphragm is severely paretic or paralyzed, however, the flaccid diaphragm cannot counterbalance the negative changes in pleural pressure generated by contraction of the inspiratory muscles of the neck and rib cage. Instead of moving normally in a caudad direction the flaccid diaphragm moves paradoxically cephalad into the thorax. This change in diaphragm motion gives rise to a paradoxical inward motion of the upper abdomen indicative of severe diaphragm weakness or paralysis.
Changes in rib cage and abdominal pressure, or volume displacement during respiration, can provide important information about diaphragm strength. Partitioning of respiration can be examined from changes in abdominal and pleural pressures, as proposed by Macklem et al.38 Changes in abdominal and pleural pressures during inspiration, expressed as the ratio of delta Pab:delta PPL are normally negative as pleural pressure becomes more negative and abdominal pressure becomes more positive. This ratio has a maximum value of +1 when the diaphragm does not contribute to inspiration and is valid only if the expiratory muscles do not contribute significantly to the pressures being generated. Alternatively, the partitioning of ventilation can be noninvasively measured by compartmental changes in rib cage and abdominal volume by respiratory inductance plethysmography or magnetometry. Optoelectronic plethysmography (OEP) is a technique that allows for measurement of chest wall and abdominal motion as well as lung volumes. A series of 89 retroreflective markers are placed on the thorax and abdomen that are monitored by a series of six to eight cameras (Fig. 84-8). The cameras record the distance the markers move in space and using Gauss’ theorem the total thoracoabdominal lung volume can be calculated. OEP can differentiate between volume generated by the rib cage (rib cage musculature) and volume generated by the abdomen (diaphragm).39 OEP can also be used to measure flow and is a nonvolitional test that can be done in any spontaneously breathing individual without regard to the degree of muscle weakness.39 This technique has been used in patients with DMD to show that with disease progression the proportion of the tidal volume attributable to the abdomen decreases while the tidal volume attributable to the rib cage increases. Furthermore those that had this change in breathing pattern were more likely to have nocturnal desaturation.40
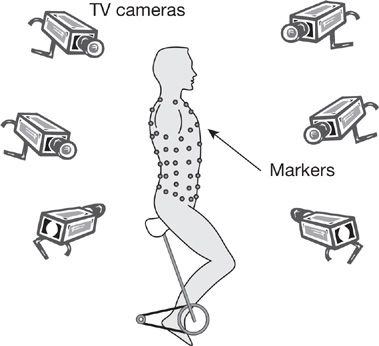
Figure 84-8 An example of optoelectronic plethysmography. A series of markers are placed on the subject’s thorax and abdomen. A group of cameras record the distance the markers move in all planes thus permitting calculation of lung volumes during the breathing cycle. (Reproduced with permission from Vogiatzis et al. Eur J Appl Physiol. 2005; 93(5–6):581–587.)
 SPIROMETRY
SPIROMETRY
Respiratory muscle weakness induced by neuromuscular disease produces a restrictive pattern on spirometric testing with a reduction in VC.6 As previously mentioned, the reduced VC is commonly out of proportion to the reduction in maximal respiratory muscle force. Reductions in lung and chest wall compliance also probably contribute. Moreover, because of the contour of the pressure– volume curve, large reductions in the respiratory muscle forces have to occur before VC is significantly reduced. A decrease in VC greater than 25% on moving from the upright to supine postures has been used as a sign of diaphragmatic weakness and a greater likelihood of sleep-related hypoventilation.14,41
Forced expiratory volume in 1 second (FEV1) and measurements of midexpiratory flow rates (FEF25–75 or FEF50) are often greater than normal predicted values in patients with neuromuscular disease. Further increases in expiratory flow may occur in patients with neuromuscular disease due to increased lung recoil. Two independent studies have shown that partial curarization in normal subjects produces a decrease in peak expiratory flow with an increase in midexpiratory flow rates compared to baseline.3,42 Moreover, myasthenia gravis patients at their baseline weakness prior to the administration of pyridostigmine have increased midexpiratory flow rates over the range of VC when referenced to absolute lung volumes (Fig. 84-9).6
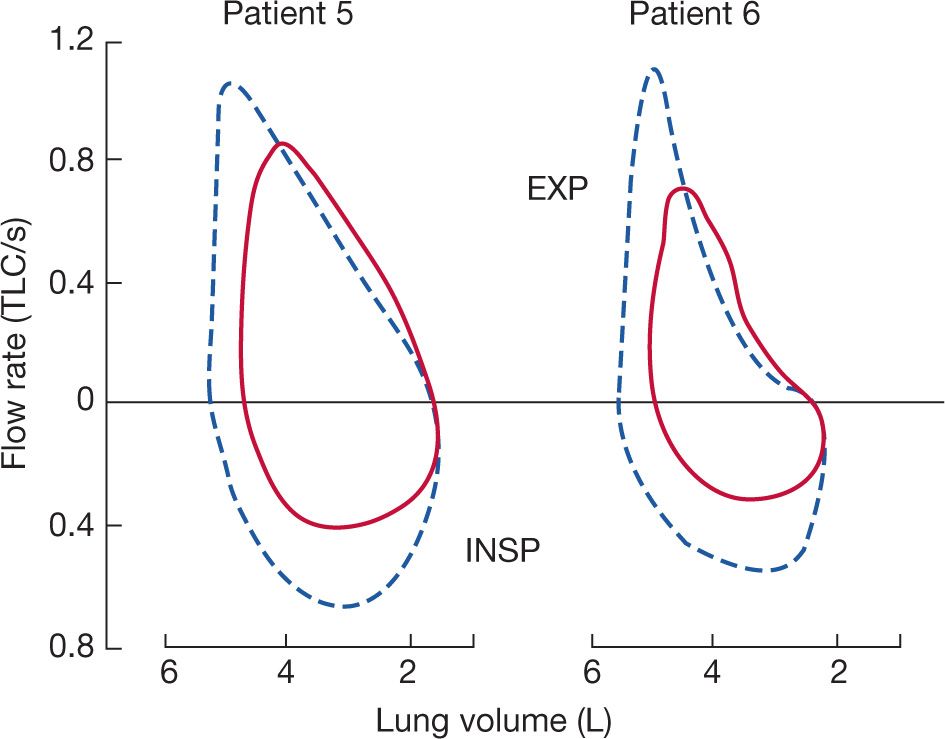
Figure 84-9 Two representative patients with myasthenia gravis and respiratory muscle weakness illustrating the effect of anticholinesterase therapy on maximum flow–volume curves. Solid curves represent pretreatment data; dashed curves were obtained following the injection of pyridostigmine. Prior to administration of pyridostigmine the midexpiratory flow rates were greater even though respiratory muscles were weaker and lung volumes were lower. (Reproduced with permission from DeTroyer A, Borenstien S. Acute changes in respiratory mechanics after pyridostigmine injection in patients with myasthenia gravis. Am Rev Respir Dis. 1980;121(4):629–638.)
 FLOW–VOLUME LOOPS
FLOW–VOLUME LOOPS
Changes in the configuration of the flow–volume loop occur in various neuromuscular diseases.2,43,44 These changes reflect respiratory muscle weakness or malfunction of upper airway muscles. “Sawtoothing” of the flow contour is seen in extrapyramidal disorders affecting upper airway muscles.45 Similarly, plateauing of the inspiratory flow waveform indicative of extrathoracic airway obstruction has been described in vocal cord paralysis caused by extrapyramidal neuromuscular disorders. An abnormal flow–volume curve is significantly more common in patients with clinically apparent bulbar muscle involvement (90% vs. 15%, respectively), and the presence of an abnormal, flow–volume loop predicted bulbar and upper muscle involvement by a neuromuscular disease with a high sensitivity and specificity.46 A characteristic flow–volume contour showing involvement of the upper airway muscles by motor neuron disease is shown in Figure 84-10.
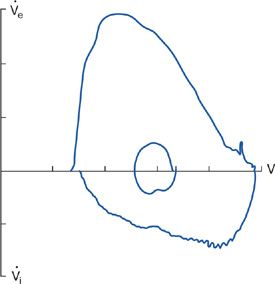
Figure 84-10 Flow–volume loop in a patient with motor neuron disease, showing inspiratory flow oscillation and inspiratory flow limitation. Subdivisions on volume and flow axis represents 1 L, flow axis 1 L/s. (Data from Vincken W, Elleker MG, Cosio MG. Determinants of respiratory muscle weakness in stable chronic neuromuscular disorders. Am J Med. 1987;82:53–58.)
Among patients with stable, chronic neuromuscular disease, the flow–volume loop is significantly more disturbed in those with respiratory muscle weakness, and these abnormalities correlate with reduced mouth pressures. Several features of flow–volume loop configuration correlate with reduced maximum static inspiratory and expiratory mouth pressures; a reduced peak expiratory flow, decreased slope of the ascending limb of the maximum expiratory curve, a drop-off of forced expiratory flow near RV, and a reduction in forced inspiratory flow at 50% of VC (Fig. 84-11). A flow–volume loop score composed of the aforementioned parameters has a high degree of specificity and 90% sensitivity in predicting respiratory muscle weakness.46
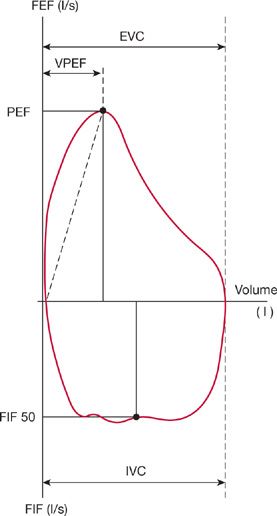
Figure 84-11 Representative flow–volume loop of a patient with chronic neuromuscular disease, showing different volume loop parameters indicative of respiratory muscle weakness. These parameters quantify the effects of respiratory muscle strength on the effort-dependent portions of the flow–volume loop. These four parameters are peak expiratory flow (PEF); ratio of PEF to the exhaled volume at which PEF was achieved, rapid vertical drop of forced expiratory flow at residual volume, and forced midinspiratory flow. (Data from Vincken W, Elleker MG, Cosio MG. Determinants of respiratory muscle weakness in stable chronic neuromuscular disorders. Am J Med. 1987;82:53–58.)
 LUNG VOLUMES
LUNG VOLUMES
A restrictive ventilatory pattern is demonstrated in patients with neuromuscular disease. A reduced TLC and a normal or reduced FRC are common. The RV is usually elevated and is a sign of expiratory muscle weakness.
 MAXIMUM VOLUNTARY VENTILATION
MAXIMUM VOLUNTARY VENTILATION
Maximum voluntary ventilation (MVV) is an index of respiratory muscle endurance in the presence of normal expiratory flow rates. This appears to be appropriate in patients with neuromuscular disease, since airway resistance and FRC are usually within the normal range. Values for MVV correlate with respiratory muscle strength and may be even more sensitive than VC in detecting respiratory muscle weakness.2
SELECTED NEUROMUSCULAR DISEASES
A helpful approach toward understanding how specific neuromuscular diseases affect the respiratory system is to localize the anatomic involvement of the respiratory system. A detailed description of the neuroanatomy of respiration is outside the scope of this chapter and is covered elsewhere in this text. In general, however, neuromuscular disorders can be broken down into disorders that involve the upper motor neuron, the lower motor neuron, or the muscle itself.
Lesions that arise in the cerebral cortex, brainstem, or spinal cord are classified as upper motor neuron lesions and are characterized by an increase in muscle tone or spasticity, the presence of an extensor plantar response, and increased reflex activity. Lesions in the lower motor neuron system demonstrate flaccidity, depressed reflexes, muscular fasciculations, and atrophy. The location and character of the patient’s weakness may enable one to identify the exact site of the lesion in the lower motor neuron system (i.e., the anterior horn cell, the peripheral nerve, the neuromuscular junction, or the muscle itself).
The following describes the effect of specific neuromuscular disease on the respiratory system and makes recommendations for treatment.
 UPPER MOTOR NEURON LESIONS
UPPER MOTOR NEURON LESIONS
A variety of disorders in which the primary pathology is centered on upper motor neurons have significant respiratory consequences. Each is discussed below.
Stroke
Hemispheric ischemic strokes reduce chest wall and diaphragm movement on the side contralateral to the cerebral insult. Decreased diaphragm excursion with stroke correlates with diaphragmatic cortical representation identified by transcranial magnetic stimulation. Bilateral hemispheric strokes are also associated with Cheyne–Stokes respiration, which is progressive hyperventilation alternating with hypoventilation and ending in apnea. This breathing pattern may result from increased responsiveness to carbon dioxide as result of interruption of normal cortical inhibition. The significance of Cheyne–Stokes respiration to stroke remains unclear but appears to be more common with bilateral than unilateral insults. Besides its effects on an alteration of breathing pattern, up to 50% of patients with strokes may have signs of pulmonary aspiration due to dysfunction of upper airway muscles that protect the airway.
Spinal cord injury
The degree of respiratory impairment depends on the level and extent of the spinal cord injury. High cervical cord lesions (C1–C3) cause paralysis of the diaphragmatic, intercostal, scalene, and abdominal muscles. Because all respiratory muscle activity is lost except for accessory and bulbar muscle function, high cervical cord injuries almost always require ventilatory assistance. In some patients, spontaneous breathing can be accomplished by glossopharyngeal breathing or diaphragmatic pacing because the phrenic nerve motor neurons (C3–C5) remain intact.
Middle cervical cord (C3–C5) lesions destroy the phrenic motor neurons and prohibit the use of phrenic nerve pacing. Patients with more caudal lesions (i.e., C4–C5 level) have an improved chance to wean from ventilator support compared to those with more cranial lesions (40% of patients with C3 lesions remain ventilator dependent.). Patients with lower cervical (C6–C8) and upper thoracic (T1–T6) cord lesions have intact diaphragm and neck accessory muscle action, but have denervated intercostal and abdominal muscles. These patients usually require ventilatory support only during the period immediately after the injury and rarely require long-term ventilation. Despite this these patients still have increased mortality. In a group of spinal cord injury patients not requiring mechanical ventilation or tracheostomy followed prospectively for a median of 4.5 years, predictors of death included age, cardiac disease, diabetes, smoking history, and lower FEV1.47 The decline in FEV1 and FVC in these patients is related to aging, increasing BMI, smoking, persistent wheeze, and lower MIP.48 These data suggest that there are modifiable risk factors that can be altered to improve outcomes in those with spinal cord injuries that do not require mechanical ventilation.
In a study of C5 or lower spinal cord–injured patients, inspiratory muscle strength was reduced to approximately 60% of predicted but was dependent on the level of cord injury. In this study, PImax values in low cervical, midthoracic, and lower thoracic–upper lumbar lesions were 61%, 69%, and 75% of predicted, respectively, whereas PEmax values were 30%, 32%, and 54% of predicted, respectively. The lower PEmax values were explained by a paralysis of abdominal and intercostal muscles resulting in reduced cough and decreased clearance of bronchial secretions. Abdominal muscle paralysis probably accounts for an abnormally compliant abdomen in patients with lower spinal cord injury, which is in stark contrast to the 30% reduction in chest wall compliance believed due to abnormal rib cage stiffness.8
Patients with spinal cord injuries also have alterations in thoracoabdominal motion during tidal breathing that is further accentuated by changing from the erect to supine position. In quadriplegic patients with relatively intact diaphragm function, the distribution of respiratory muscle weakness results in paradoxical inward motion of the upper rib cage during inspiration owing to weakness of the parasternal and scalene muscles. This pattern of abnormal thoracoabdominal movement is more marked in the supine than in the upright position. Patients with high quadriplegia (above C3–C5) may be able to sustain short periods of spontaneous respiration because of inspiratory activity of the sternocleidomastoid and trapezius muscles. Phasic inspiratory EMG activity has been observed in the platysma, mylohyoid, and sternohyoid muscles. Analysis of ribcage motion in these patients shows increased upper rib cage diameter, due to the inspiratory action of the neck accessory muscles pulling the sternum cranially and expanding the upper rib cage.
The distribution of muscle paralysis in low cervical cord spinal patients also has a profound effect on the performance of forced expiratory maneuvers. In contrast to healthy normal subjects, in whom VC is moderately decreased on assuming the supine position, in quadriplegic patients there is a paradoxical increase in VC in the supine compared to seated position without a significant increase in TLC. In 14 quadriplegic patients (C4–C7), there was a 16% increase in VC on changing from the upright to supine position and a reduction in RV (29%) and TLC (on average, 6%).27 The mechanism believed to be responsible for the increase in VC in supine quadriplegic patients is the hydrostatic effect of abdominal contents, causing cephalad displacement and diaphragm lengthening and thereby placing the diaphragm on a more favorable portion of its length–tension curve. The use of elastic binders when quadriplegics assume upright posture has been advocated to prevent the increase in abdominal compliance. Abdominal binding may have physiologic benefit by maintaining diaphragm precontraction length in a more optimum position on its length–tension curve.49
It was previously believed that all expiratory muscles were paralyzed in lower cervical cord injuries. However, studies of C5 to C8 quadriplegics indicate that phasic EMG activity of the clavicular portion of the pectoralis major is associated with a marked decrease in the anteroposterior diameter of the upper rib cage.50 This portion of the pectoralis muscle receives innervation from the C5 to C6 cord level. With the arms placed at the subject’s side, contraction of the caudate head of the pectoralis major causes caudal displacement of the manubrium sterni and upper rib cage. This expiratory action has been shown to decrease expiratory reserve volume (ERV) by 60% when the shoulders are held in abduction. After 6 weeks of pectoralis muscle isometric training, patients with low quadriplegia can have a marked increase in maximum pectoralis muscle isometric strength and a significant reduction in ERV.51 Conceivably, therefore, training of this muscle could improve the effectiveness of cough in patients with low spinal cord injury.
In the months following spinal cord injury, pulmonary function typically improves. In patients with spinal injuries below the C5 level, VC is approximately 30% of predicted in the first week after injury, but increases to 45% of predicted by the fifth week and by the fifth month to approximately 60% of predicted.52 Improvements in VC have been attributed to spasticity developing in previously flaccid intercostal and abdominal muscles thereby increasing the rigidity of the thorax and abdomen and improving diaphragm force generation.
There is a role for corticosteroid use in the acute management of spinal cord injury. Methylprednisolone given as a bolus 30 mg/kg followed by a 24-hour infusion at 5.4 mg/kg/h has been shown to improve motor function at 6 weeks, 6 months, and 1 year, but only in those received the drug within 8 hours of injury.53 A subsequent study compared methylprednisolone infusion (5.4 mg/kg/h) for 48 hours to 24 hours after the administration of a bolus (30 mg/kg).54 There was no difference in functional outcome between the two infusion periods except in those where the bolus dose was given between 3 to 8 hours after the injury. If the methylprednisolone was started between 3 to 8 hours after the injury, then those that received the infusion for 48 hours did have improved motor function at 6 weeks and 6 months. There were higher rates of pneumonia and sepsis in the 48-hour infusion group but mortality was not different.54 No trial has shown a mortality benefit, and it should be recognized that the outcome measured was an improvement in the functional independence measure (FIM) score and not a return to normal motor function.
Parkinson Disease
Parkinson disease is due to degeneration of neurons in the substantia nigra and has a prevalence in the United States of approximately 200 cases per 100,000 people. Parkinson disease can be either primary (e.g., idiopathic) or secondary, as in postencephalitic parkinsonism associated with the influenza pandemic, or part of a more generalized disorder, such as multiple system atrophy or drug abuse with MPTP (1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine).
Parkinson disease has been thought to be a purely motor disorder but recently non-motor findings are being associated with Parkinson disease, which can predate motor symptoms.55 These findings have led to speculation that Parkinson disease could alter respiratory control at the level of the brainstem. A study in 15 subjects with early Parkinson disease and normal respiratory flow and volumes found that 7/15 had an abnormal ventilatory response while 11/15 has an abnormal occlusion pressure response (P0.1) to hypercapnic challenge testing suggesting abnormal respiratory control.56
Respiratory abnormalities are common in Parkinson disease, with pneumonia being the most common cause of death. A substantial problem with Parkinson disease is glottic muscle dysfunction.57 An abnormal flow–volume loop contour showing flow oscillations commonly occurs. On direct fiberoptic visualization of the upper airway, these oscillations correspond to rhythmic involuntary movements of glottic and subglottic structures. Physiologic evidence of upper airway obstruction may be present. In addition to the presence of oscillations in flow, a rounding off of the peak of the midexpiratory flow–volume curve, a lowered peak expiratory flow rate, and a delayed appearance of peak expiratory flow have been observed in Parkinson patients. These results have been interpreted as evidence for less coordinated or less “explosive” respiratory muscle contractions.43,45
Patients with mild-to-moderate Parkinson disease are able to perform simple single respiratory efforts (e.g., measurements of lung volume and maximum static inspiratory pressures) but have difficulty performing more complex, repetitive ventilatory efforts (i.e., sustaining inspiratory resistive loads to exhaustion and performing maximum unloaded breathing efforts). Performance of repetitive respiratory tasks is associated with an increased work of breathing when compared to that of an age-matched control group. These findings are similar to derangements in task performance exhibited by peripheral skeletal muscle groups in Parkinson patients.26
Treatment significantly improves neurologic scores, maximum expiratory pressures, and peak inspiratory flow. A recent meta-analysis has demonstrated that levodopa improves peak expiratory flow and FVC in Parkinson disease.28 Deep brain stimulation by stereotactically placing electrodes into the suprathalamic nucleus or the globus pallidus nucleus has been shown to be equally effective when treating medically resistant patients.58 The electrodes produce a low-voltage high-frequency stimulation that results in inhibition of the neurons in the nucleus. Although the effect on respiratory function has not been directly studied, this procedure has been shown to improve motor function and quality of life.59
Multiple Sclerosis
MS is a demyelinating disorder of the central nervous system, characterized clinically by remissions and relapses of clinical symptoms due to disseminating CNS lesions. MS is the most common neurologic disease afflicting young adults, with an estimated prevalence of 250,000 to 300,000 cases in the United States in 1990. The cause of the disease is unknown, although epidemiologic evidence points to genetic and environmental factors. Classic clinical symptoms include paresthesia, motor weakness, diplopia, blurred vision, dysarthria, bladder incontinence, and ataxia.
Because MS can cause focal lesions anywhere in the central nervous system, different patterns of respiratory impairment can occur. Impairment of the respiratory centers and the medulla can cause failure of automatic breathing (Ondine’s curse), apneustic or neurogenic pulmonary edema. The three most common respiratory manifestations of MS are respiratory muscle weakness, bulbar dysfunction, and abnormalities in respiratory control.
Acute respiratory failure rarely occurs in this disease, but it can occur because of severe demyelination of the cervical cord. Diaphragmatic paralysis resulting in respiratory insufficiency has also been reported. Even with severe disability and impaired respiratory muscle strength, patients with MS seldom complain of dyspnea. This paucity of respiratory complaints may be due to restricted motor activities and greater expiratory than inspiratory muscle dysfunction. Clinical signs that may be helpful in predicting respiratory muscle impairment are weak cough and inability to clear secretions, limited ability to count on a single exhalation, and upper extremity involvement. Advanced MS is frequently complicated by aspiration, atelectasis, and pneumonia.
In a group of 38 patients that were not bedridden or wheelchair bound without bulbar involvement and a diagnosis of MS for 9.2 years, there was a significant decrease in the maximal inspiratory pressure (MIP) and the maximal expiratory pressure (MEP) to 77% and 60% predicted respectively.60 In a group of 21 ambulatory stable MS patients, the percent predicted MEP was significantly reduced when compared to age-matched healthy controls (69.4% vs. 85.6%, p = 0.03). Furthermore, these patients had a greater change in the upright versus supine FVC compared to healthy controls (262 vs. 98 mL, p = 0.001).61 Not surprisingly the MEP had an inverse correlation (r = –0.47; p = 0.04) with MS functional scores. These data suggest that even in ambulatory MS patients without respiratory symptoms respiratory muscle weakness is present, and should be monitored periodically (particularly MEP). In 60 bedridden MS patients, pulmonary function studies revealed severely decreased MIP (47% predicted), MEP (30% predicted), and VC that was 80% of predicted. In those with a VC below 80% predicted, the MIP and MEP were significantly lower than those with a normal VC.62 In all of these studies the MEP was more affected than the MIP, and the respiratory muscle weakness directly correlated with the severity of the subject’s overall neurologic function. Smeltzer et al. developed a pulmonary dysfunction index for patients with MS and found that it is correlated with MEP measurements. The score assesses the patient’s assessment of cough and ability to handle secretions, the examiner’s assessment of cough, and how high the patient can count on a single exhalation (Table 84-6). A subject with normal cough efficacy would have a score of 4 while an individual with the most impairment would have a score of 11.63 Gosselink et al.29 examined the effect of respiratory muscle training (e.g., three sets of 15 expiratory contractions at 60% of MEP twice daily) on respiratory muscle strength and the subject’s pulmonary index score in a group of severe MS patients. At 3 months there was a statistically significant improvement in MIP, and although the MEP improved the p value was 0.07 compared to control patients. The pulmonary index was statistically better at 3 months and 6 months following expiratory muscle training but the FVC was not.29 A subsequent study in 17 ambulatory MS patients who underwent expiratory muscle training was able to statistically improve MEP but this did not improve pulmonary function or markers of cough effectiveness.64



