The Genetic Basis of Respiratory Disorders
The field of genetics and genomics is advancing at an incredible pace. The completion of the Human Genome Project was just the beginning. Now, thanks to rapid advances in sequencing technology and bioinformatics, we have made significant progress toward sequencing 1000 genomes from around the world, uncovering great genetic diversity and challenging us to understand the biologic relevance. Sequencing the exome (the protein-coding parts of the genome) of a patient with an undiagnosed condition is already a reality. Even beyond human genetics, genomic technologies are making an impact on pulmonary disease, enabling characterization of new respiratory pathogen genomes, such as the SARS virus and pandemic H1N1 influenza, with unprecedented speed. Against this backdrop, a chapter on the genetics of lung diseases could easily be out of date before it is even in print. Thus it does not seek to be encyclopedic, but to give the reader a grounding in the principles of human genetics, an overview of current knowledge in Mendelian lung diseases and a summary of recent progress in understanding genetic factors contributing to common lung conditions. It outlines some of the emerging roles of epigenetic modifications and aims to give a vision of where the field is moving, concluding with current and future prospects for genetically targeted therapies.
PRINCIPLES OF HUMAN GENETICS
 GENOME ORGANIZATION
GENOME ORGANIZATION
The term “genome” refers to the genetic make-up of an organism (Table 7-1). Mammalian genomes are composed of deoxyribonucleic acid (DNA) and can be subdivided into a nuclear genome – DNA within the nucleus of each cell – and a separate circular genome housed within each mitochondrion. DNA has a double-helix structure, each strand comprises four constituent bases – adenine (A), cytosine (C), guanine (G), and thymine (T) – that pair together, A with T and G with C. DNA needs to be replicated each time a cell divides. This strict base pairing ensures accurate copying of the DNA code.
TABLE 7-1 Glossary of Genetic Terms
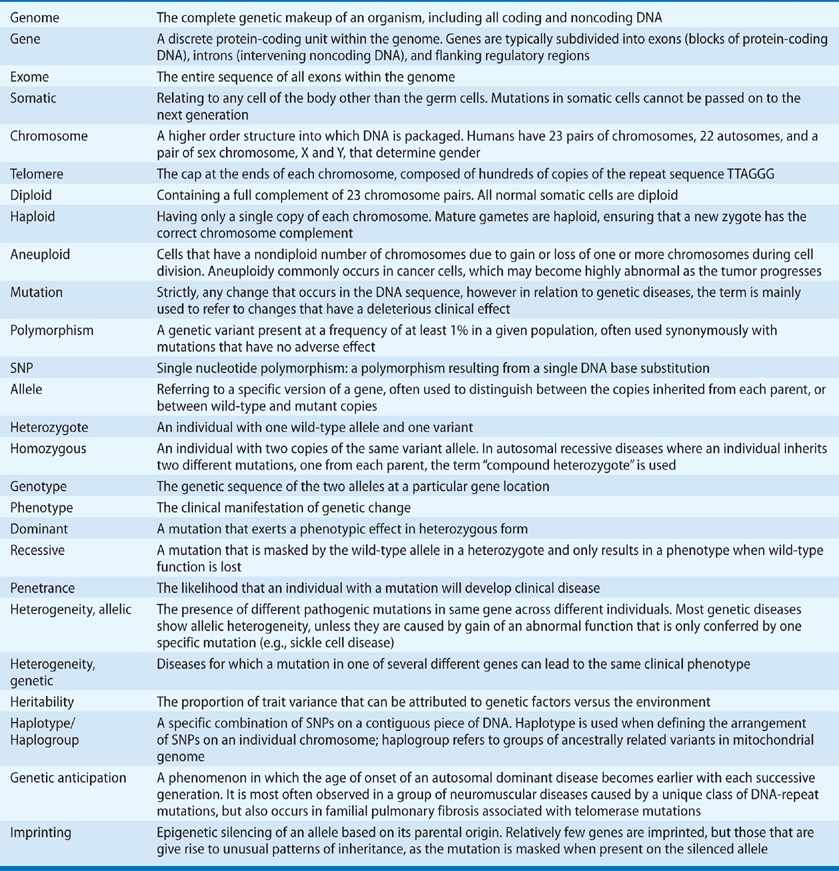
The human genome is approximately 3.3 billion base pairs in size. This large amount of DNA is wound around proteins known as histones, then packaged into higher-order structures called chromosomes that can be visualized under a light microscope. Most cells of the body contain two copies of each chromosome; one inherited from each parent, and are termed “diploid.” Diploid cells contain 23 pairs of chromosomes. During meiosis, these pairs of chromosomes are separated, giving rise to oocytes and sperm that contain a single copy of each chromosome (termed “haploid”). Thus fusion of two haploid gametes gives rise to a new diploid organism, preserving the correct copy number of DNA through the generations. Gender is determined by a pair of sex chromosomes, X and Y; females have two copies of the X-chromosome, whereas males have one each of X and Y. The other 22 pairs of chromosomes are known as the autosomes. Genetic diseases are mainly caused by mutations in autosomal or X-chromosome genes; the Y-chromosome harbors only a few genes and these are mainly involved in determining male characteristics.
 GENE STRUCTURE
GENE STRUCTURE
Only about 1% to 2% of the human genome actually encodes for proteins. The noncoding portion was originally considered to be junk DNA, but it is now increasingly clear that some of it has important regulatory functions. The protein-coding units are called genes. The DNA within a gene is first transcribed into ribonucleic acid (RNA). RNA has a similar base structure to DNA, but is single stranded, has a slightly different sugar backbone and thymine (T) is replaced by uracil (U). Genes are typically divided into coding exons and intervening noncoding introns. The intronic sequences are spliced out of the initial RNA transcript to produce the mature messenger RNA (mRNA) molecule. Some genes have alternative splicing patterns that can give rise to slightly different variants (“isoforms”) of the protein. The mRNA is then translated into protein by ribosomes. Ribosomes read the RNA code as a triplet of bases or “codon” and add the corresponding amino acid to the growing protein chain. There is some redundancy in the genetic code and amino acids may be encoded by several different codons. Four codons have a special function: AUG encodes methionine and always marks the initiation site for protein translation, while UGA, UAG, and UAA are stop signals that lead to termination of translation. The DNA flanking the coding region of a gene is not translated but contains important regulatory elements, including the promoter region that regulates transcriptional activity.
 CLASSES OF MUTATION
CLASSES OF MUTATION
Alterations in the DNA sequence occur when there is an error in DNA replication prior to cell division or DNA damage occurs through environmental exposures such as UV radiation or tobacco smoke. Such mutations may affect a single base (known as a point mutation) or may involve the insertion or deletion of multiple bases. Cells have an extensive DNA repair mechanism that will correct most of these mutations, but any that escape may lead to a permanent change in the sequence that is propagated to daughter cells. Mutations in noncoding regions of the genome often have no detrimental effect and over time, they may become quite common in the population. Variants that are present at a frequency of greater than 1% are known as polymorphisms and have been widely used in genetic mapping studies. Thus the two copies of a gene in any individual are subtly different at the DNA sequence level. These variant forms are known as “alleles.” However, mutations that occur in the introns of genes may lead to disease, especially if they disrupt the highly conserved splicing signals immediately flanking an exon. Within the coding region of a gene, the consequence of a point mutation depends on whether it alters the genetic code (Fig. 7-1). Most redundancy lies in the third base of the codon, so for example, a change from GGG to any of GGA, GGC, or GGU still encodes glycine and would not change the sequence of the protein. Such changes are usually silent and may become common polymorphisms. Mutations that lead to an amino acid substitution, for example UGU (cysteine) to UAU (tyrosine), are known as missense mutations. Their effect on protein function depends much on the specific structure and function of that protein. In general, missense mutations in regions that are functionally critical, such as the catalytic domain of an enzyme, will be highly deleterious. These regions are often highly conserved across species, indicating that mutations have not been tolerated during evolution. Missense mutations that affect residues important in secondary structure and protein folding are also likely to have adverse effects, whereas mutations in linker regions may be less critical. Thus interpreting the consequences of genetic changes requires an in-depth knowledge of the protein concerned.

Figure 7-1 Classes of genetic mutations. A. Single base changes within an exon may lead to premature protein truncation (nonsense mutation, shaded in red), an amino acid substitution (missense mutation, yellow), or there may be no change due to redundancy in the genetic code (silent, blue); B. Insertions or deletions lead to frameshift mutations if the number of bases involved is not divisible by three. This almost invariably leads to premature protein truncation downstream of the mutation site.
Mutations that lead to premature truncation of a protein are highly likely to be pathogenic and are a major cause of inherited diseases. Several different types of mutation can lead to premature protein truncation. Nonsense mutations result from a single base change that introduces a stop codon earlier than the natural translation end-point, for example, AGA (arginine) to UGA (stop). Small insertions and deletions can also introduce premature stop codons because if the number of bases added or lost is not a multiple of three, the reading frame for the triplet codon is offset and it is read incorrectly. This is known as a frameshift mutation (Fig. 7-1). Splice-site mutations can lead to retention of an intron, which does not normally code for a protein and, therefore, often contains a stop codon. Alternatively splice-site mutations can lead to exon skipping and again, if the size of the missing exon is not a multiple of three bases, this leads to a downstream frameshift, in addition to losing a whole exon of sequence. The presence of a premature stop codon often triggers a process known as nonsense-mediated mRNA decay (NMD), which leads to degradation of the nonsense-containing mRNA transcript, preventing translation of a truncated protein. This protects the cell from potentially adverse effects of an abnormal protein product. The mechanisms underlying NMD are not fully understood and some transcripts are degraded more efficiently than others. Nonsense mutations in the last exon of a gene do not trigger NMD, due to their proximity to the natural stop codon, whereas mutations in first exon, close to the translation initiation codon, may lead to reinitiation at a downstream ATG site.
The last major class of mutation is gene rearrangements, large deletions, or duplications that affect one or more exons. These mutations can be missed by sequence-based methods of DNA analysis and require specialized methods that measure the copy number of DNA across the gene. They typically lead to major disruption of the gene structure and any protein that may be produced is likely to be nonfunctional.
 MODES OF INHERITANCE
MODES OF INHERITANCE
The pattern of inheritance of a genetic disease within a family is determined by the location of the mutation – on an autosome, the X-chromosome, or in the mitochondrial genome – and whether or not a clinical effect (phenotype) is evident when only one copy of the gene is mutated. When mutation of a single allele is sufficient to cause disease, it is known as dominant because the mutation is sufficient to overcome the positive effect of the remaining wild-type allele. Genes that are affected by dominant mutations are typically very sensitive to the 50% reduction in gene dosage that results from inactivating one allele. Alternatively the mutation may cause an abnormal gain of function, or create an abnormal protein that in turn interferes with the function of the wild-type protein, an effect known as dominant negative. In contrast, recessive mutations have no detrimental effect when only one allele is mutated. The remaining wild-type allele is sufficient to maintain normal gene function and a clinical phenotype is only apparent when both alleles are inactivated and the gene function is completely lost. Individuals with a personal or family history of genetic disease should be offered genetic counseling to help them understand their risks and options, and to facilitate appropriate genetic testing.
 AUTOSOMAL DOMINANT
AUTOSOMAL DOMINANT
Autosomal dominant mutations result in a strong pattern of disease in each generation of a family (Fig. 7-2A). An individual with such a mutation has a 50% chance of passing the disease on to each of their children. The hallmarks of autosomal dominant inheritance are approximately equal proportions of males and females affected by the disease and the presence of male-to-male transmission. However, several factors can complicate this model. Some dominant diseases may skip a generation, due to reduced penetrance (Fig. 7-2B). The penetrance of a mutation is defined as the likelihood that someone with the mutation actually develops the disease. If the penetrance is less than 100%, then an individual who inherits the mutation may escape the disease themselves, while still being at 50% risk of passing it on to their children. Independently, the gender ratio may skewed by environmental and/or genetic modifying factors. The most extreme example is inherited diseases affecting sex-specific organs. For example, ovarian cancer can be inherited in an autosomal dominant manner but only females with the mutation actually develop the disease.
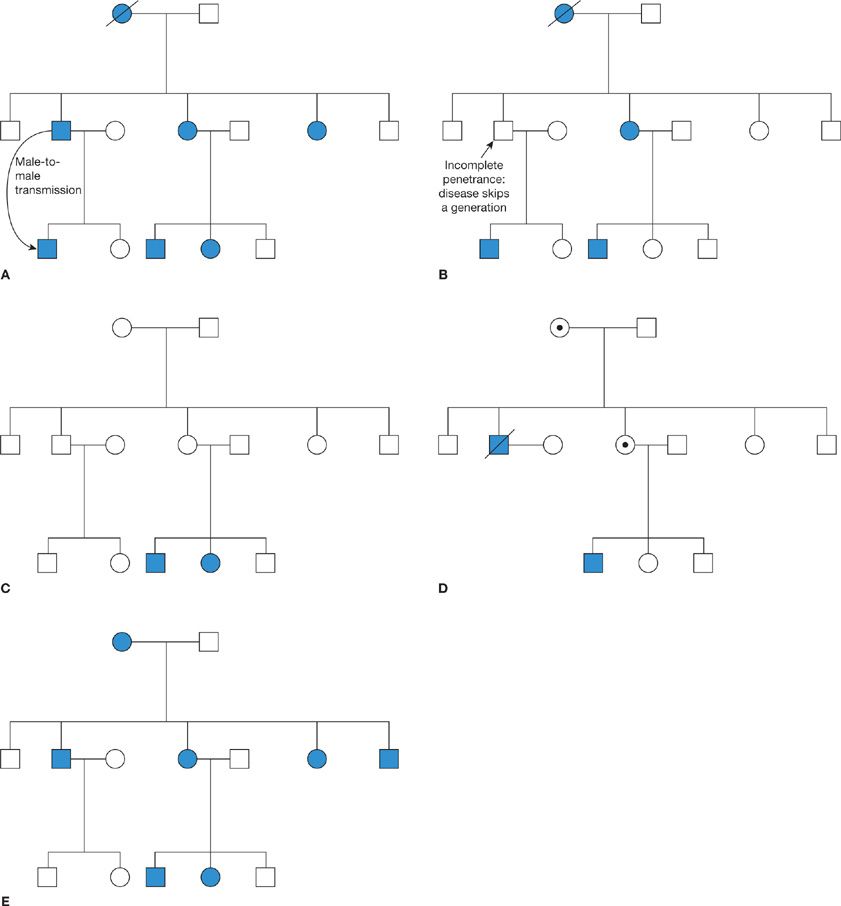
Figure 7-2 Patterns of inheritance of genetic diseases. A. Autosomal dominant inheritance is characterized by the presence of disease in every generation, equal gender distribution, and male-to-male transmission; B. When an autosomal dominant disease has reduced penetrance, it may skip a generation; C. Autosomal recessive disease suddenly appears in the family when both parents are heterozygous mutation carriers; D. X-linked recessive diseases are transmitted by carrier females but in general, only males are affected. Male-to-male transmission is impossible; E. Mitochondrial diseases are transmitted through the female lineage and can potentially affect all children born to an affected mother. Key: Squares denote males; circles, females; solid symbols denote individuals affected by the disease; circles with a dot in the center denote obligate carriers of an X-linked condition.
 AUTOSOMAL RECESSIVE
AUTOSOMAL RECESSIVE
The pattern of inheritance of autosomal recessive diseases is quite different. Individuals with a single copy of the mutation are known as carriers or heterozygotes. Since there is no clinical effect, such individuals are usually unaware of their status. However, when both parents are carriers, each has a 50% chance of passing on the mutation, meaning there is a 25% chance that a child will inherit two copies of the mutation and be affected by the disease. Consequently, autosomal recessive diseases often appear “out of the blue” in a family with no previous history of the condition (Fig. 7-2C). The incidence of recessive diseases largely depends on the heterozygote frequency in the population, but the risk increases in consanguineous families.
 X-LINKED INHERITANCE
X-LINKED INHERITANCE
X-linked diseases are caused by mutations in genes on the X-chromosome. Most are recessive, but the different sex-chromosome constitution between females and males, XX versus XY, leads to a unique pattern of inheritance. Males who inherit an X-linked mutation have no wild-type allele on the Y-chromosome to mask its effect, and consequently they develop the disease. Female carriers are generally unaffected, as for autosomal recessive diseases. Thus X-linked diseases are passed through the female line and typically affect only males (Fig. 7-2D). A female carrier has a 50% chance of an affected son. Importantly, males cannot pass the mutation to their sons, so an evidence of male-to-male transmission rules out X-linked inheritance, but all of their daughters will be carriers. In reality, females only have a single X-chromosome active in any given cell, due to a process of X-inactivation in early embryonic development that adjusts the dosage of X-linked genes to be the same as in males. If X-inactivation is random, approximately half of cells in a carrier will express the wild-type allele and half express the mutation, meaning that female carriers usually have no phenotype or are only mildly affected. However, if X-inactivation is highly skewed toward expression of the mutant allele, then female carriers may be as severely affected as males. Rarely, X-linked mutations may be dominant, meaning that all females who inherit the mutation will be affected. Such mutations are often lethal in males. Examples of X-linked recessive conditions that affect the lung include X-linked agammaglobulinemia, an immunodeficiency that can lead to chronic lung disease, and X-linked severe combined immunodeficiency caused by mutations in the IL2RG gene that encodes a subunit of the receptor for multiple interleukins.
 MITOCHONDRIAL MUTATIONS
MITOCHONDRIAL MUTATIONS
The mitochondrial genome is a small circular molecule, approximately 16,500 bases long. It encodes some of the proteins required for oxidative phosphorylation and electron transport, together with multiple transfer RNAs and ribosomal RNAs. Mutations in mitochondrial genes adversely affect energy production and thus the clinical consequences are greatest in tissues with high-energy requirements, such as heart, brain, and skeletal muscle. Two characteristics make the inheritance of mitochondrial gene mutations unique. First, mitochondria are almost exclusively transmitted through the maternal lineage; sperm only have mitochondria in the tail for motility and do not enter the oocyte at fertilization. Thus, the pattern of inheritance within a family is similar to X-linked inheritance, with no male-to-male transmission, but differs in that females and males are equally likely to be affected (Fig. 7-2E). In theory, all children born to an affected mother would inherit the mutation and develop the disease. In reality, however, there are many copies of the mitochondrial genome per cell and each cell has a mixture of wild-type and mutant mitochondria (heteroplasmy). The segregation of these mitochondria during cell division is random, so by chance, an oocyte may have a high or low number of mitochondria carrying the mutation. This random drift continues throughout embryonic development and beyond, generating considerable variability in the severity of disease and the tissues that are affected, even among individuals in the same family. Pulmonary involvement is not a major feature of most mitochondrial diseases, but several case reports link pulmonary hypertension with mutation in mitochondrial genes or a nuclear-encoded mitochondrial protein.1–8 Pulmonary complications of mitochondrial disease are most likely to present as part of a multiorgan syndrome that may also include cardiac and/or skeletal myopathy, neuropathy, retinopathy, renal problems, or metabolic abnormalities.
Another fascinating property of the mitochondrial genome is its high degree of polymorphic variation. Clusters of variants, or “haplogroups,” have been used to plot early human migration patterns across the globe. As new variants arose, they were propagated to offspring in the immediate geographic area, but were not present in other populations that had already migrated to different regions. Some of these variants confer subtle functional differences and may have been selectively enriched by helping adaptation to a new environment. They may also modulate risk of disease, particularly for conditions where there is oxidative stress. Data concerning lung diseases are currently limited, but associations with different haplogroups have been reported for atopy and asthma, chronic obstructive pulmonary disease (COPD), high-altitude pulmonary edema, and lung cancer risk.9–13
SOMATIC MUTATIONS AND CANCER
Not all of the genetic changes that contribute to disease are inherited. This is particularly true in cancer where, although there may be an inherited predisposition, most genetic changes are somatic and confined to the tumor itself. A later chapter is devoted to the molecular basis of lung cancer, so here we will briefly review the types of somatic changes observed in cancer cells and their relevance to benign lung conditions.
Two major classes of genes may be mutated in cancer: oncogenes and tumor suppressor genes. Oncogenes promote tumorigenesis when they are expressed at an abnormally high level or are inappropriately expressed in tissues where the gene should normally be silent. This may occur due to amplification (extra copies) of the gene, overactivation by upstream transcription factors, a chromosome rearrangement that brings the gene under the control of a strong promoter, or loss of DNA methylation as described in the following section on epigenetics. Alternatively the gene may be mutated in a way that gives the protein a novel gain of function. For example, mutation of a ligand-dependent receptor such that, once activated, it cannot be switched off and continues to signal in the absence of the ligand. These types of oncogenic mutations are usually dominant missense mutations at specific amino acid sites within the protein.
Tumor suppressor genes (TSGs) are like the brakes on the cell; they control cell growth, differentiation, and apoptosis. When their function is lost, the cell proliferates uncontrollably or evades programmed cell death. TSGs are predominantly inactivated by nonsense and frameshift mutations, large gene deletions, or loss of an entire chromosome. They may also be silenced by hypermethylation of their promoter, as described in the next section. In contrast to oncogenes, TSG mutations are often recessive at the cellular level and both copies of the gene must be inactivated before the full cancer-promoting effect is seen. Both mutations may occur as somatic changes in the cell that initiates the cancer, or the first mutation may be inherited, predisposing the individual to the risk of cancer, a model that was first proposed by Alfred Knudson.14
As tumors proliferate, their genome may become highly disorganized. Abnormal segregation of the chromosomes during mitosis can lead to aneuploidy, with gains and/or losses of entire chromosomes. There may also be translocations, where segments of different chromosomes are inappropriately joined together, and localized deletions or duplications of large segments of DNA. Such large rearrangements will clearly affect many different genes and can contribute to the activation of oncogenes and/or loss of TSG function.
The study of somatic mutations requires tissue from the affected area, ideally with a comparison to normal tissue from the same patient and also normal tissue from unrelated controls. Due to the difficulty of obtaining such tissues for benign lung diseases, somatic changes have mainly been studied in the context of cancer, but the same approach has recently been applied to pulmonary arterial hypertension (PAH). In addition, somatic epigenetic changes described below are common both in cancer and in several chronic lung diseases, emphasizing the importance of acquiring tissue from the site of disease when this is ethically possible.
EPIGENETICS
 DNA AND HISTONE MODIFICATIONS
DNA AND HISTONE MODIFICATIONS
The term epigenetics refers to factors that influence gene expression without altering the underlying base sequence. Both DNA and histones, the proteins around which DNA is wound, may be epigenetically modified. These changes are usually reversible and play important roles in regulating gene expression and genome stability. The most common DNA modification is methylation of cytosine residues. The promoters of many genes contain a CpG island, a region with a high density of CG dinucleotides. Methylation of CpG sites in these islands leads to a closed chromatin conformation that makes the DNA inaccessible to transcription factors, turning off expression of the gene. Conversely, when most of the cytosines are unmethylated, the DNA is open and actively transcribed. DNA methylation, therefore, plays a critical role in regulating tissue-specific patterns of gene expression. Patterns of DNA methylation are controlled by DNA methyltransferases (DNMTs). DNMT3A and 3B are responsible for de novo methylation of residues that were previously unmethylated. Established patterns of methylation are then maintained by DNMT1. Further fine tuning of gene regulation comes through methylation and acetylation of histones. Acetylation mainly occurs on lysine residues and relaxes the interaction between histone and DNA, leading to increased gene transcription. Deacetylation reverses this and leads to a more tightly closed chromatin conformation. Histone acetylation patterns are controlled by histone acetyltransferases (HATs) and deacetylases (HDACs).
Within noncoding regions of the genome, DNA methylation and chromatin condensation act to suppress repetitive elements that could otherwise recombine and cause structural alterations. In cancer, there is often a global loss of methylation, which can lead to activation of mobile and repetitive elements, predisposing to the genomic instability that is the hallmark of many cancers. Loss of methylation at gene promoters can also activate oncogenes that in turn accelerate the growth of the tumor. At the same time, there may be hypermethylation of specific gene promoters, causing loss of expression of TSGs. It is increasingly clear that more subtle epigenetic changes likely contribute many other diseases, including lung conditions such as idiopathic pulmonary fibrosis (IPF) and COPD.15,16 Unlike the DNA sequence, epigenetic modifications can change dynamically with age and are influenced by dietary factors such as folate intake. There is also mounting evidence that airborne pollutants such as small diesel particulates and tobacco smoke can directly mediate epigenetic changes.17,18 Thus the lung may be particularly susceptible to epigenetic changes caused by repeated exposure to these environmental modulators. Importantly, though, some adverse epigenetic changes are reversible with time, for example, smoking-induced changes in DNA methylation gradually revert after quitting.19 Also, the anti-inflammatory action of corticosteroids is in part epigenetic, recruiting HDAC2 to the site of acetylated (activated) inflammatory genes.20 Characterizing the role of epigenetics in lung disease is challenging because it requires access to affected and control lung tissues. It may also be difficult to distinguish which changes are causative of disease and not just a reaction to the disease state. However, considerable progress has already been made under the auspices of the NIH Roadmap Epigenomics Consortium and other focused research initiatives.
 NONCODING RNAs
NONCODING RNAs
Noncoding RNAs can directly regulate gene expression at the RNA level without being translated into a protein product. The best characterized family is the microRNAs (miRs), first studied in plants but now also recognized to be important throughout the animal kingdom. Primary miR transcripts are transcribed in the same manner as regular protein-coding genes. In some cases the miR gene may be within an intron of a protein-coding gene and is controlled by the promoter of the “parent” gene. In other cases, miRs may be encoded as separate genes, individually or in a cluster, with their own promoter. The primary miR transcript is then processed into a pre-miR, about 70 to 80 nucleotides in length (Fig. 7-3). The ends of the pre-miR are highly homologous, causing the molecule to loop back on itself in a hairpin-like conformation. This double-stranded RNA structure is then exported from the nucleus into the cytosol, where it is cleaved by the enzyme Dicer into a mature single-stranded miR, approximately 18 to 22 nucleotides long (Fig. 7-3). The mature miR negatively regulates gene expression by binding to the 3′-untranslated region of its target mRNA, which either leads to degradation of the mRNA or inhibits protein synthesis. The seed sequence that initiates binding between the miR and its mRNA target is very short, typically around seven nucleotides, and does not require perfect base pairing. As a consequence, a single miR can potentially target tens or even hundreds of genes.
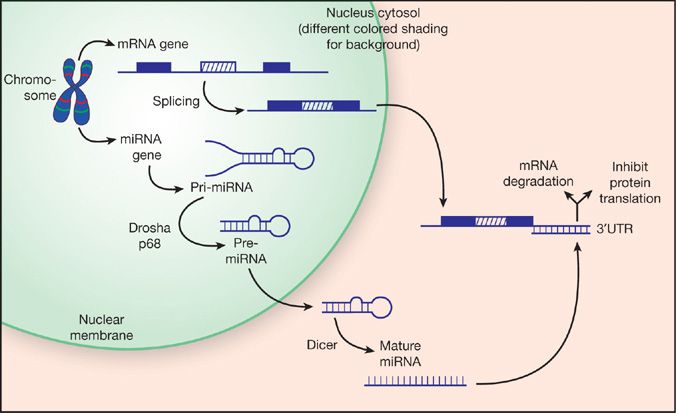
Figure 7-3 microRNA biogenesis. microRNAs that are encoded by independent genes are transcribed by RNA polymerases (mainly RNA pol II) into a primary miRNA transcript with a 5′-cap and 3′-polyadenlyation. The primary miRNA molecule is cleaved by a protein complex, including Drosha and p68, into a double-strand hairpin RNA known as the pre-miRNA. For a subset of microRNAs, recruitment to the p68-Drosha complex is stimulated by activation of the bone morphogenetic protein and transforming growth factor-beta pathways,67,68 a process that is disrupted by some mutations that cause pulmonary arterial hypertension.58 Alternatively, some miRNA genes are embedded within the introns of mRNA genes, in which case they are transcribed along with the host gene and the pre-miRNA is generated during mRNA splicing. The pre-miRNA is then exported to the cytosol, where the Dicer complex converts it to the mature single-stranded miRNA molecule. Mature microRNAs negatively regulate expression of their target genes by binding to the 3′-untranslated region of the mRNAs, which either leads to degradation of the mRNA or blocks protein translation.
miRs have been widely studied in cancer and several, such as miR-21, have been dubbed “oncomiRs” because their overexpression promotes tumorigenesis by downregulating tumor suppressor pathways.21–24 In comparison, relatively little is known about the role of miRs in benign lung diseases. However, it is now an intense area of research and recent data highlight important roles in IPF and PAH, as detailed in the disease-specific sections that follow. The miR pathway is often considered to be an epigenetic mechanism, yet it can be influenced by changes in DNA sequence, either through mutation affecting the miR gene itself, or mutations and polymorphisms that alter the seed sequence in the mRNA target.
Another major class of noncoding RNAs is the long noncoding (lnc) RNAs, greater than 200 bases long. lncRNA transcripts can be thousands of bases in length and, unlike miRs, they may be encoded by large multiexon genes that undergo splicing in the same manner as most protein-coding genes. Well-known examples of lncRNAs are XIST, which coats the inactive X-chromosome in female cells, and TERC, part of the telomerase complex that maintains the ends of the chromosomes (telomeres). As yet, very little is known about the role of lncRNAs in human disease, the notable exception being TERC mutations, one of several causes of IPF described below.
INHERITED LUNG DISEASES
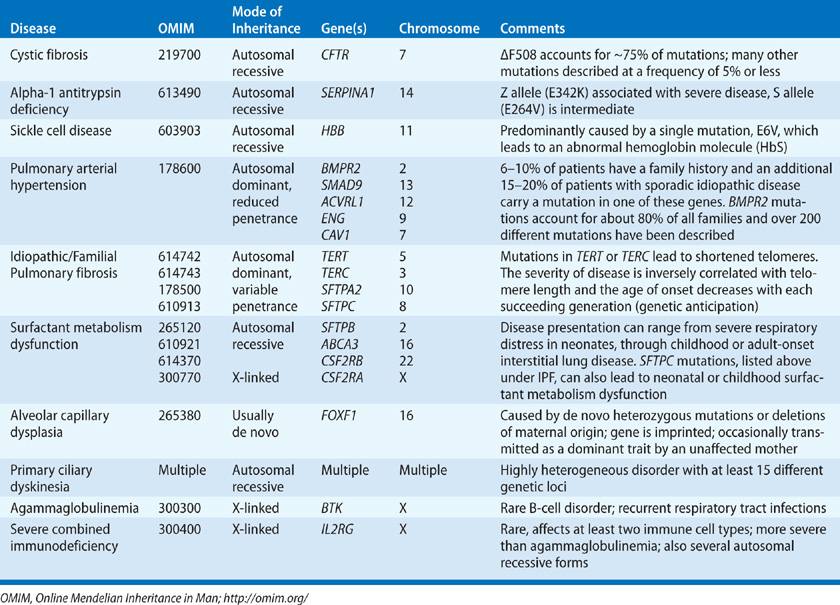
This section provides an overview of the genetic basis of inherited conditions that include lung disease as a major component (Table 7-2). These brief summaries cannot provide exhaustive reviews of current knowledge, but we refer the reader to entries in Online Mendelian Inheritance in Man (OMIM) and other web resources listed at the end of this chapter, for in-depth information.
TABLE 7-2 Mendelian Inherited Lung Diseases
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


