The Eosinophilic Pneumonias
INTRODUCTION
The association between pulmonary infiltrates and eosinophilia was first identified by Loeffler in 1932. It is now recognized that the eosinophilic pneumonias are a heterogeneous group of disorders characterized by varying degrees of pulmonary parenchymal and/or blood eosinophilia.1 The precise role that eosinophils play in the pathogenesis of the different eosinophilic pneumonias is not clear. Normally, less than 2% of the leukocyte cell differential in bronchoalveolar lavage (BAL) are eosinophils. The presence of increased BAL and/or tissue eosinophils, and our knowledge of the biology of eosinophils (see Chapter 22) does, however, suggest that they play a variety of roles, including initiation, perpetuation, and amplification of tissue inflammation and injury. These effector functions are no doubt the result of the ability of the eosinophils to release numerous soluble mediators, including granule-derived proteins, arachidonic acid metabolites, proinflammatory cytokines, superoxide anions, metalloproteases, and hydroxyl radicals. The different roles of eosinophils in these disorders can be appreciated when comparisons are made of parasitic infections and disorders such as asthma or allergic bronchopulmonary aspergillosis (ABPA). In the former, eosinophils play a crucial role in eradicating the infectious pathogen; in the latter, the eosinophils accumulate in the lung as a result of immune hypersensitivity and are prominent mediators of tissue injury.
The spectrum of diseases that can be primarily or secondarily associated with blood or pulmonary eosinophilia is shown in Table 71-1. It is beyond the scope of this chapter to discuss each of these disease entities in detail. Instead, discussion will focus on diseases of known or unknown causes in which eosinophilic infiltration of lung tissue is a characteristic feature, including acute eosinophilic pneumonias, tropical pulmonary eosinophilia (TPE), chronic eosinophilic pneumonia (CEP), ABPA, Churg–Strauss syndrome (now termed as eosinophilic granulomatosis with polyangiitis [EGPA]), and idiopathic hypereosinophilic syndrome (HES). Since eosinophilic granuloma of the lung is frequently seen in the absence of blood or tissue eosinophilia, it is considered separately (Chapter 74).
EOSINOPHILIC PNEUMONIAS WITH ACUTE PRESENTATIONS
Acute presentations of eosinophilic pneumonia center on several primary considerations, including Loeffler Syndrome, parasitic infections, drug- and toxin-related disorders, and idiopathic varieties. Each is discussed in the following sections.
 LOEFFLER SYNDROME (SIMPLE PULMONARY EOSINOPHILIA)
LOEFFLER SYNDROME (SIMPLE PULMONARY EOSINOPHILIA)
In 1932, Loeffler first described a clinical syndrome characterized by mild respiratory symptoms, peripheral blood eosinophilia, and transient, migratory pulmonary infiltrates. The term Loeffler syndrome, or simple pulmonary eosinophilia, has been used to define the numerous similar cases reported subsequently. Immune hypersensitivity to Ascaris lumbricoides has been recognized as the likely cause of most of the earliest reported cases, although several other parasitic infections, including hookworms (Ancylostoma duodenale, Necator Americanus, Necator Brasilensis), Strongyloides, Trichinella Spiralis, and Toxocara Canis2–4 and exposures to numerous drugs and other agents have also been recognized to induce a Loeffler-like syndrome (see below and Tables 71-2 and 71-3). An identifiable etiologic agent may be lacking in up to one-third of patients.
aDrugs commonly or occasionally reported to cause pulmonary eosinophilia.
Loeffler syndrome affects people of all ages. It is characterized clinically by the presence of low-grade fever, nonproductive cough, dyspnea (mild to severe), chest discomfort with coughing or deep breathing, and, occasionally, hemoptysis.3,5 The respiratory manifestations of Loeffler syndrome are usually self-limited, typically resolving in 1 to 2 weeks. Laboratory examination of peripheral blood from patients reveals moderate-to-extreme eosinophilia, which may be at peak levels as respiratory symptoms resolve and which resolves over several weeks.4 Expectorated sputum, if present, frequently contains eosinophils and/or Charcot–Leyden crystals.4,5 Transient, migratory, nonsegmental, bilateral, interstitial, and alveolar infiltrates (often peripheral or pleural based) are evident on the chest radiograph.3 Infiltrates typically clear after several weeks. Pulmonary function evaluation typically reveals a mild-to-moderate restrictive ventilatory defect with a reduced diffusing capacity for carbon monoxide (DLCO).
When Loeffler syndrome is due to A. Lumbricoides, hookworms or other parasites, the pulmonary manifestations are believed to result from a hypersensitivity reaction to the parasite larvae. Following ingestion of Ascaris ova, larvae hatch within the small intestine, then cross the intestinal wall to enter the splanchnic, and ultimately the pulmonary circulation. Subsequently, the larvae migrate across pulmonary capillaries into alveoli, mature into adult worms, ascend the large airways, and are swallowed into the gastrointestinal (GI) tract, where they complete their life cycle. The pulmonary manifestations of Loeffler syndrome begin approximately 9 to 14 days following ingestion and occur during the migration of larvae through the lung. Ascaris suum, a large roundworm endemic to pigs, can cause a nearly identical syndrome. Cutaneous penetration of larvae is the principal relevant mode of tissue entry for hookworms.
During the pneumonic stage of the illness, Ascaris or hookworm larvae may be identified in sputum or gastric aspirates.4 In keeping with the life cycle of Ascaris or hookworms, stool examination for ova and parasites is typically negative until 8 weeks after the onset of the respiratory syndrome.6 Histologic evaluation of lung tissue is not required for confirmation of the diagnosis. When tissue has been obtained, a characteristic and striking eosinophilic infiltration of interstitium and alveolar–capillary units has been noted. Increased numbers of macrophages have also been appreciated. Tissue necrosis and vasculitis are not features of the disorder. Ascaris or hookworm larvae may be identified in the tissue specimen.4
Since Loeffler syndrome may be induced by a variety of exposures, a search for an etiologic agent (e.g., parasitic infection or drug reaction) should be undertaken. Bronchodilators and rarely corticosteroids may be used for alleviation of pulmonary symptoms, although these are usually self-limited. In cases due to Ascaris, treatment with oral mebendazole (100 mg twice a day for 3 days or a single dose of 500 mg) should be given to prevent late GI manifestations of Ascaris infestation, which may include malnutrition, diarrhea, abdominal pain, and/or intestinal obstruction typically 8 weeks or more after onset of respiratory symptoms. Pyrantel pamoate, albendazole, or ivermectin are alternate treatment options.4 Since stool specimens are negative for ova and parasites early in the illness, clinical follow-up over a 2- to 3-month period is indicated.
 PARASITIC INFECTIONS
PARASITIC INFECTIONS
Infections with parasites other than Ascaris species are also commonly associated with pulmonary infiltrates and blood or pulmonary eosinophilia.3–5 The parasites associated with the development of pulmonary eosinophilic syndromes are listed in Table 71-2. The prevalence of infection with each of these organisms varies with geographical location, socioeconomic status, and host immunity. Parasites may infect the lung via direct pulmonary invasion or via hematogenous seeding. In addition to Ascaris species, Strongyloides stercoralis (an intestinal nematode), Ancylostoma brasiliensis (cutaneous helminthiasis, “creeping eruption”), Ancylostoma duodenale, and T. canis (dog roundworm, “visceral larva migrans”) are the parasitic agents most commonly associated with pulmonary eosinophilia in the United States.
Strongyloides is widely distributed in the tropical and subtropical regions.3 Following initial transcutaneous infection, a Loeffler-like syndrome may occur as larvae migrate through the lungs. Chronic strongyloidiasis occurs as a result of autoinfection, whereby the noninfectious rhabditiform larvae transform within the GI tract into infectious filariform larvae, penetrate the colonic wall or perianal skin, and reinfect the host.4,5 Chronic strongyloidiasis may be associated with recurrent asthma-like symptoms that may worsen with the administration of corticosteroids. The hyperinfection syndrome results from accelerated autoinfection, and usually occurs in persons with defects in cell-mediated immunity3 (such as lymphoma, human immunodeficiency virus [HIV] or human-T lymphotropic virus type 1 [HTLV-1] infection, and with chronic corticosteroid use), as well as in persons with underlying GI disease, chronic lung disease, malnutrition, and use of H2 blockers or antacids.7 It may also occur in healthy persons. Respiratory manifestations include cough, dyspnea, chronic bronchitis, wheezing, hemoptysis, and patchy pulmonary infiltrates, in association with blood eosinophilia. Rarely, acute respiratory distress syndrome (ARDS) has been reported in patients with hyperinfection. GI manifestations are also common, including abdominal pain, paralytic ileus, nausea and vomiting, bowel perforation, and secondary sepsis from gram-negative bacteria. Central nervous system (CNS) manifestations such as meningitis have also been noted.
The diagnosis of Strongyloides infection may be established by identification of larvae in sputum, BAL fluid, bronchial brushings, or transbronchial biopsy specimens, pleural fluid or stool. Several stool samples are often required to identify the pathogen.5 Serologic testing, such as ELISA to detect IgG antibody to Strongyloides stercoralis can also be used to establish a diagnosis.4 Patients at risk for Strongyloides hyperinfection syndrome should be screened for the parasite prior to initiation of immunosuppressive therapy.3
Thiabendazole (25 mg/kg twice a day for 2 days) or ivermectin (200 μg/kg given orally for 1–2 days) may be used for the treatment of uncomplicated or disseminated strongyloidias.4 Ivermectin is generally better tolerated in terms of side effects.5 Albendazole is an alternative agent. Higher dose and longer duration of thiabendizole treatment are needed to treat disseminated strongyloidiasis in immunocompromised persons.5 The hyperinfection syndrome associated with Strongyloides can be difficult to cure. Therapy should be continued until the clinical syndrome resolves and larvae are no longer detectable in the GI tract.
Ancylostomiasis is a nematodal infection endemic to the southeastern coastal regions of the United States, Mexico, and Central and South America.4,5 The organism is present in soil contaminated by stool from infected domestic animals. It penetrates human skin most commonly through the feet. This results in the development of the “creeping eruption” lesion – a raised, erythematous, serpiginous, tunnel-like, and often itchy lesion on areas of exposed skin.8 A Loeffler-like syndrome occurs in up to 50% of cases of “creeping eruption.” Specific treatment for pulmonary involvement is typically not required as illness is usually self-limited.
Infection with T. canis may occur throughout the world and leads to the clinical syndrome of “visceral larva migrans.”4 This syndrome is characterized by hepatomegaly, leukocytosis, fever, hypergammaglobulinemia, and persistent blood eosinophilia.9 Because the disease most commonly affects young children, a high degree of clinical suspicion is necessary to establish the diagnosis in adults. Respiratory symptoms, including cough and severe wheezing, may occur after ingestion of substantial numbers of larvae. Laboratory evaluation reveals peripheral blood and BAL eosinophilia, elevated serum levels of immunoglobulin E (IgE), and poorly defined, diffuse nodular alveolar infiltrates on chest radiograph.4 ELISA testing for larval antigens is diagnostic. Although the disease may be self-limited, treatment with thiabendazole, albendazole, mebendazole, diethylcarbamazine, or corticosteroids may hasten recovery in patients who are severely ill.3–5
 DRUG AND TOXIN-INDUCED PULMONARY EOSINOPHILIC SYNDROMES
DRUG AND TOXIN-INDUCED PULMONARY EOSINOPHILIC SYNDROMES
A vast number of drugs and toxic exposures have been associated with the development of pulmonary infiltrates and blood or pulmonary eosinophilia.1,10–13 A partial list of these medications and exposures is given in Table 71-3, and information regarding pulmonary drug toxicities may also be found on the Internet on the regularly updated web site, www.pneumotox.com. Of the medications implicated, many are commonly used antibiotics, nonsteroidal anti-inflammatory agents, anticonvulsants, cardiovascular medications, and antidepressants.
In addition to medications, a number of toxic exposures may also be associated with eosinophilic pneumonia.10 For example, eosinophilic pneumonia has been described following radiation therapy for breast cancer, dust or smoke exposure,14–17 exposure to iodinated contrast agents or 1,1,1-trichloroethane (Scotchguard),18 and after inhalation of cocaine or heroin.19–21
Whereas most cases of drug- or toxin-induced pulmonary eosinophilia are sporadic, outbreaks of pulmonary eosinophilia have occurred following ingestion of rapeseed oil (contaminated with aniline derivatives)22 or L-tryptophan.23 The precise incidence of drug- or toxin-induced pulmonary eosinophilia is difficult to assess, since most of the literature pertaining to these syndromes is published in the form of case reports, rather than large series or controlled trials. For the same reason, the precise pathogenesis and the definition of the clinical syndromes associated with individual exposures are difficult to characterize.
In general, drug-induced pulmonary eosinophilic syndromes have an acute or subacute onset and are not always related to either the cumulative dose of drug used or the duration of treatment. Respiratory symptoms vary widely in severity, from a mild Loeffler-like illness with dyspnea, cough, and fever to severe fulminant respiratory failure. The DRESS syndrome consists of acute eosinophilic pneumonia with drug rash and systemic manifestations.24 Wheezing may be present, but obstructive physiology is not common on pulmonary function testing. Although radiographic findings are not specific, interstitial or alveolar infiltrates are typically evident on chest radiograph (Fig. 71-1), and common high-resolution chest computed tomographic (CT) findings include bilateral consolidation and ground-glass opacities, both of which are frequently peripherally located.
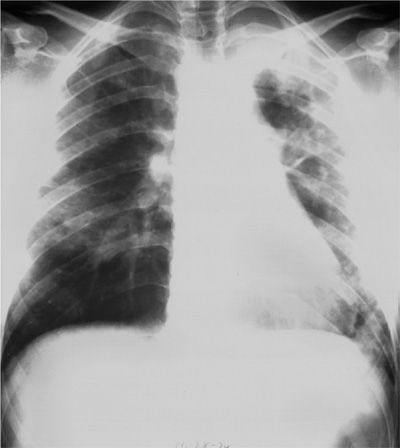
Figure 71-1 Chest radiograph of a 23-year-old woman with acute sulfasalazine-induced eosinophilic pneumonia. Bilateral interstitial and alveolar infiltrates are present.
A diagnosis of drug- or toxin-induced eosinophilic pneumonia is based upon a careful review of drug and other exposures (including nonprescription drugs, herbal preparations, street drugs, and environmental exposures). Other causes of eosinophilic lung disease must be excluded. A concurrent skin rash and pleural effusion can support the diagnosis of drug-induced eosinophilic pneumonia. In some cases, testing with lymphocyte proliferation assays may reveal T-cell sensitization to specific drugs. However, the utility of such assays is limited as negative tests do not rule out a drug-induced disorder, and these assays are not widely available for routine clinical use. The prognosis is favorable in most cases. Elimination of exposure to the drug or other toxin usually leads to resolution of symptoms, eosinophilia, pulmonary infiltrates, and normalization of lung function within a month. Supplemental therapy with corticosteroids is not universally required, but it may hasten recovery in patients who are severely ill.
 IDIOPATHIC ACUTE EOSINOPHILIC PNEUMONIA
IDIOPATHIC ACUTE EOSINOPHILIC PNEUMONIA
In contrast to the typically benign Loeffler syndrome, a more severe idiopathic form of eosinophilic pneumonia termed acute eosinophilic pneumonia (AEP) has been recognized as a distinct clinical entity.25–28 Although seen in patients of both genders and any age group, AEP tends to occur in patients between the age of 20 and 4026,29,30 and is more common in men.25 AEP is usually idiopathic, occurs commonly in previously healthy persons, and may represent an acute hypersensitivity reaction to an inhaled agent.28 Similar cases have been reported in persons with a history of chronic myelogenous leukemia, hematopoietic stem cell transplantation31,32 or HIV infection.33 Many cases have been reported in patients who have recently commenced smoking,27,34–36 used flavored tobacco products,37 or had other changes in smoking habits. The disease may recur when former smokers resume smoking.35,38 Overall up to 70% of patients with AEP have a history of smoking.26,39 In addition, cases have been reported in persons treated with venlafexine,40 minocycline,26 daptomycin,11,41 and several other drugs.26 It has also been reported among persons who have been involved in activities with unusual exposures (including exposure to dust from the World Trade Center collapse in New York City,14 after military deployment in Iraq,17 cave exploration, gasoline tank cleaning, plant repotting, woodpile moving, and indoor renovations).26,42 AEP has also been seen following inhalation of cocaine or heroin21,43 and in association with H1N1 influenza infection.1,44 Although none of the patients in the original reported series had atopy or asthma, cases have since been described in persons with a history of atopy. No definite seasonal variation has been identified.
Idiopathic AEP presents as an acute illness with dyspnea, fever, nonproductive cough, tachypnea, pleuritic chest pain, and hypoxemia (arterial PaO2 under 60 mm Hg) at times with myalgias.25,26,39 Symptom duration is typically less than 7 days,1 although longer courses of up to 30 days have been described. Patients usually have diffuse inspiratory crackles on chest auscultation, wheezing may be present, and rapid progression from mild dyspnea to overt respiratory failure requiring mechanical ventilation is common.17,26,27 A moderate leukocytosis with left shift is typical,27 but blood eosinophilia is usually absent at the onset of disease.17,25,26 Early clinical features may be mistaken for community-acquired pneumonia. Blood eosinophilia may develop later in the course of the disease and may provide a clue to the diagnosis.27,45 Serum IgE levels may be moderately elevated.26,46 The erythrocyte sedimentation rate (ESR) may be elevated as well. Serum levels of thymus and activation-regulated chemokine (TARC)/CCL17 (a ligand for CCR4 on Th2 lymphocytes) may be elevated and may help to distinguish AEP from other cause of acute lung injury(ALI).47 Striking eosinophilia (25%–55%) is present in BAL fluid.17,26,27,45,48 Increased numbers of lymphocytes (up to 20%) and neutrophils (up to 15%) are commonly also present in BAL fluid in AEP.48 Pulmonary function tests reveal a restrictive ventilatory defect with a reduced DLCO that typically normalize following treatment.48
Early in the course of illness, the chest radiograph reveals subtle, patchy infiltrates with Kerley B lines.26,49 Diffuse, symmetric alveolar and interstitial infiltrates resembling ARDS with a ground-glass or micronodular or reticular appearance (Fig. 71-2A) develop within 48 hours.50,51 Infiltrates are typically bilateral, although AEP with unilateral infiltrates has been described. Small-to-moderate bilateral pleural effusions are common (affecting up to 50%–70% of patients).27 Fluid analysis typically reveals a high pH and marked eosinophilia.51 CT scanning confirms the presence of diffuse parenchymal ground-glass attenuation, inerlobular septal thickening and/or consolidation (Fig. 71-2B), with prominence along bronchovascular bundles, with or without pleural effusion.49,52 Lymphadenopathy may also be seen.
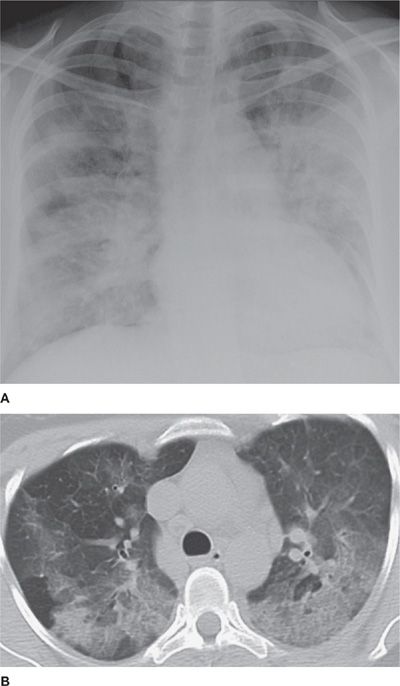
Figure 71-2 Radiographic apœpearance of idiopathic acute eosinophilic pneumonia (AEP). A. Diffuse bilateral alveolar and interstitial infiltrates apparent on chest radiograph. B. Diffuse parenchymal ground-glass opacity and consolidation evident on computed tomography scan.
Light microscopic examination of lung tissue reveals prominent eosinophil infiltration in interstitium and/or alveolar spaces, and bronchial walls.53 The pathologic pattern of diffuse alveolar damage with hyaline membranes and eosinophilic infiltrates should suggest the possibility of AEP. Lymphocytic infiltration of interstitium, type 2 pneumocyte hyperplasia, and intra-alveolar fibrinous exudate are also common. Granulomas, alveolar hemmorhage, and nonnecrotic perivascular inflammation have been reported.53 Basal lamina damage is unusual.49 Extrapulmonary involvement is rare.
The pathogenesis of idiopathic AEP is poorly understood.26,39 The occurrence of cases following unusual environmental exposures (as noted earlier) suggests these exposures as possible disease-inciting events, perhaps as triggers for a hypersensitivity reaction to an unidentified antigen in susceptible persons. Of note, elevated levels of the fungal cell wall component β-D-glucan have been described in the BAL fluid of some patients with AEP, suggesting a possible association between exposure to fungus and development of disease.
However, the roles of lymphocytes and eosinophils in this disorder have not been fully elucidated. Elevated levels of interleukin (IL)-5, a Th2 lymphocyte–derived cytokine involved in activation and recruitment of eosinophils, have been described in the BAL of patients with AEP.54 Levels of vascular endothelial growth factor (VEGF), a cytokine induced by IL-5, have also been shown to be elevated in BAL and to correlate with number of eosinophils and levels of IL-5.55 Elevated BAL levels of IL-18, a cytokine capable of inducing several cytokines known to induce or enhance eosinophilia, have also been identified among patients with acute (and other) forms of eosinophilic pneumonia.26 Collectively, these findings suggest a role for Th2 lymphocytes and eosinophils in disease pathogenesis. It remains unknown, however, whether the eosinophils initiate the disease process or are a secondary manifestation of the disorder. Alveolar macrophage–derived cytokines may also play a role in the development of AEP.56
Idiopathic AEP is a diagnosis of exclusion51 and should be considered in a patient who presents with an acute febrile illness less than 1 week in duration, apparent ALI or ARDS without a typical antecedent illness. A careful search must be undertaken for other causes of pulmonary infiltrates, especially fungal or other infection, and drug or other exposures. Specimens of blood, sputum, stool, BAL, and often transbronchial biopsy specimens should be obtained for stain and culture as well as serologic testing to rule out viral, bacterial, mycobacterial, fungal, and parasitic infection.26,39 BAL cell differential should be performed. Elevated blood levels of TARC/CCL17 may distinguish AEP from other causes of ALI, even in the early phase of disease before blood esoinophilia is present.47 In contrast, levels of KL-6, a marker for alveolar cell damage, tend to be lower in AEP than in other forms of ALI.47 An elevated fraction of exhaled nitric oxide (FeNO; e.g., levels >23.5 ppb) may also help to distinguish AEP from non-AEP disorders57 and FeNO levels decreased following corticosteroid treatment. Lower serum IgG levels have also been reported in AEP as compared with other causes of pulmonary eosinophilia.58
Idiopathic AEP generally carries an excellent prognosis. Although fatalities have been reported, most patients demonstrate rapid dramatic responses to corticosteroid therapy,25,26,59 with abatement of fever and respiratory symptoms within 12 to 48 hours and complete resolution of infiltrates, pleural effusion, and pulmonary function impairment usually within 1 month.27,48 The optimal steroid regimen for the treatment of AEP has not been determined. However, initial doses of methylprednisolone typically used range from 60 to 125 mg administered every 6 hours. After resolution of respiratory failure, oral prednisone (in doses of 40–60 mg per day) may be continued for 2 to 4 weeks with a subsequent slow taper over the next several weeks.26,51 Despite the apparent clinical success of steroid treatment, there is no definitive proof that steroids alter the natural history of the disease. Spontaneous disease regression has been reported,27 and in contrast to idiopathic CEP, absence of clinical relapse is characteristic. Follow-up pulmonary function testing is generally normal, although a small number of patients may demonstrate mild reductions in DLCO or lung volumes.
TROPICAL PULMONARY EOSINOPHILIA
TPE was first described in the early 1940s60 as a syndrome characterized by fevers, malaise, anorexia, weight loss, paroxysmal dry cough with dyspnea or wheezing, marked peripheral blood eosinophilia, and spontaneous resolution over several weeks’ time. In the 1950s and 1960s, filarial infections were recognized as the cause of this disorder.61 TPE is most prominent in India, Africa, and Southeast Asia, but it may be seen worldwide in filarial-endemic regions.3,4,62 Disease may also present in nonendemic regions among immigrants or travelers.62,63 A rare manifestation of parasitic infection, TPE occurs in less than 1% of patients infected with lymphatic filariae (usually introduced by mosquito bite) and results from a hypersensitivity reaction to microfilariae from Wuchereria bancrofti and Brugia malayi.5,62,64 Illnesses resembling TPE have also been reported following infection with other parasites. Approximately four times more common in men, most patients with TPE manifest the disease between the age of 25 and 40 years,4,62 although children and older adults may also be affected. There is no known seasonal or genetic propensity to this disease, and it remains unclear why only such a small percentage of patients with filarial infection develop TPE.
Clinical manifestations of TPE develop months to years after the infection.4 The most common distinguishing symptom of TPE is spasmodic cough that usually occurs at night.62 Other typical early symptoms include low-grade fevers, weight loss, fatigue, and malaise. Dyspnea and wheezing, which can be severe, are common, and the clinical presentation may resemble status asthmaticus. Chest pain, muscle tenderness, and cardiac, pericardial, and CNS involvement have also been reported. Rarely, patients remain asymptomatic. Physical examination of patients with TPE is notable for coarse rales or rhonchi and wheezing,62 although no abnormalities are found in approximately 20% of patients. Generalized lymphadenopathy and hepatosplenomegaly, pericarditis, musculoskeletal or CNS manifestations may be present,4,62 but they are less common in adults than in children.
Laboratory findings in TPE include extreme peripheral blood eosinophilia4,5,62 (usually more than 3000 eosinophils per cubic millimeter and up to 90% of the leukocyte differential) that persists for several weeks, although the degree of eosinophilia generally does not correlate well with clinical disease severity or radiographic findings. Blood eosinophils appear degranulated and contain cytoplasmic vacuoles.62 Total serum IgE is usually elevated (more than 1000 U/mL), and the presence of high titers of filarial-specific IgE and IgG, measured by complement fixation or hemagglutination techniques, confirms the diagnosis.3,5 Hypergammaglobulinemia results from polyclonal activation of B cells.4 The ESR, circulating immune complexes, serum IgG, IgM and IgA, and complement (CH50) may be moderately elevated,4,5 and patients may also have an abnormal electrocardiogram (ECG). Eosinophils may be identified in the sputum,4,62 and, in those with active disease, BAL typically reveals intense eosinophilia (upto 50% of the differential), elevated levels of total IgE, and filarial-specific IgE, IgM, IgG and fibronectin.4,5,62 BAL may also contain IgE antibodies to B. Malayi BM 23–25 antigen4,62,65 as well as eosinophil-derived neurotoxin.5 Pleural fluid, when present, is eosinophilic and also contains elevated IgE.4 Serum α1-antitrypsin levels are reduced and return to normal with treatment.4,66 Microfilariae are not found in blood or sputum,4 and examination of stool or urine for ova and parasites is negative (although patients from endemic countries may be simultaneously infected with other parasites). In contrast, microfilariae have been identified in lung and lymph node tissue, especially when lymphadenopathy is present.
Pulmonary function test findings vary with the duration of disease: They reveal an obstructive ventilatory defect in up to 30% of patients, particularly when symptoms have been present less than 1 month. A restrictive ventilatory defect and reduced DLCO, with or without a concomitant obstructive defect, are typical of long-standing disease.3–5 Mild arterial hypoxemia may be present.62 Ill-defined, diffuse reticulonodular infiltrates with a mottled appearance primarily affecting the mid to lower lung fields are characteristic radiographic findings in TPE.3,4,62 Bronchovascular markings may be prominent and hilar adenopathy and pleural effusions have occasionally been reported.4,62 The chest radiograph may be normal at the time of presentation in as many as 20% of patients.62 In rare cases where Dirofilaria is the causative agent, the chest radiograph may reveal solitary or multiple nodules thought to represent infarcts caused by parasitic emboli. CT scanning may show mediastinal adenopathy, bronchiectasis, and areas of calcification.67
The histopathologic findings in TPE depend on the tissue examined, as well as the stage and duration of the disease.62 Studies of lung pathology have shown that the early stage of the disease (within the first 2 weeks) is characterized by histiocytic inflammation in the alveolar, interstitial, peribronchial, and perivascular spaces, with preservation of lung architecture.4 Tiny nodules may be palpable within the lung tissue. One to three months after symptom onset, eosinophilic infiltration with eosinophilic bronchopneumonia and microabscesses is present in lungs of untreated patients. Degenerating microfilariae may be present within the center of the microabscesses, and some destruction of alveolar walls may be evident. Local bronchial walls are also edematous and inflamed, with evidence of epithelial disruption. Long-standing untreated disease is associated with the presence of chronic mixed-cell inflammation (histiocytes, eosinophils, and lymphocytes) in a nodular pattern and the development of pulmonary fibrosis.4,62 Foreign body–type granulomatous lesions are often present. Lymph node biopsies may reveal degenerating microfilariae or adult worms, surrounded by aggregates of eosinophils, their granule products, and giant cells.
The clinical features of TPE are believed to result from an intense hypersensitivity reaction to microfilarial antigens of W. bancrofti and Brugia malayi. Although a broad spectrum of clinical disease may be caused by filaria, patients with TPE rarely have other systemic features of filariasis. Canine filarial forms (e.g., Dirofilaria immitis) are rarely transmitted to humans but also may be recovered from lung and lymph node specimens. Disease occurs when larvae introduced into the body via insect bites develop into mature filariae.4 The adult worms, dwelling within the lymphatics, produce microfilariae, which are then trapped in the pulmonary vasculature. The release of antigens from degenerating microfilariae leads to an intense local and systemic inflammatory response. A striking antibody and eosinophilic response, similar to that seen in peripheral blood, is also present within the lung.4,5,62 Although little is known about the precise mechanisms by which filariae are cleared in patients with TPE, both antibody-dependent mechanisms and eosinophils probably play a role.4,62 In vitro, both granulocytes and macrophages can bind microfilariae in the presence of IgG, IgE, or complement leading to the death of the organism. The finding of an intense lymphocytic- and plasma-cell infiltrate around microfilariae in tissues suggests that lymphocytes may be important for clearance of the organism. In vitro lymphocyte transformation in response to stimulation with microfilarial antigens can be demonstrated in some cases. The transcription factor NFkB68 and oxidants69 are also reported to play an important role in the inflammatory response to TPE. The precise mechanisms by which eosinophils accumulate in the lung and contribute to tissue inflammation in patients with TPE are incompletely understood. Lung (as well as blood) eosinophils appear degranulated by microscopy.62,70 Elevated levels of eosinophil-derived neurotoxin, an RNase capable of damaging the lung epithelium, have been observed in the BAL fluid of patients with TPE. IgE and eosinophil-, lymphocyte-, mast cell-, or basophil-derived products may contribute to the wheezing and airway hyperresponsiveness that can occur in this disorder.
The diagnosis of TPE is usually established on the basis of the clinical and laboratory findings described earlier including pertinent exposure history. Lung or other tissue biopsies are not typically required. The diagnostic criteria for TPE are summarized in Table 71-4.62 Biopsy of enlarged lymph nodes (e.g., scalene) may assist in establishing the diagnosis in some cases. A rapid treatment response may provide confirmatory evidence that the correct diagnosis has been made. The differential diagnosis includes Loeffler syndrome, CEP, ABPA, drug reactions, other parasitic infections, HES, and lymphangitic spread of carcinoma. In nonendemic areas, the disease may also masquerade as asthma, atypical pneumonia, sarcoidosis, EGPA, granulomatosis with polyangiitis (formerly known as Wegener’s granulomatosis, WG), or tuberculosis (TB). Diagnosis in nonendemic regions is often delayed, and a careful review of travel history and a high index of suspicion are necessary to prompt the diagnosis.
Diethylcarbamazine, a piperazine derivative used widely in the treatment of filarial infections, is the therapy of choice for TPE, typically at a dose of 6 mg/kg/d for 14 to 21 days.4,5,62 Diethylcarbamazine acts by both direct and indirect mechanisms. It is directly filaricidal to both adult worms and microfilariae. It can also enhance the binding of granulocytes, macrophages, antibodies, and complement to the surface of microfilariae. A marked clinical improvement and decrease in eosinophil count usually occurs in the first 7 to 14 days of therapy. Clinical improvement following diethylcarbamazine treatment has been correlated temporally with the resolution of eosinophilic alveolitis. In addition, improvement in pulmonary function, reduction in BAL eosinophilia, a decrease in total and filarial-specific IgE and IgG, increase in serum α1-antitrypsin levels, and radiographic clearing generally occur within 1 to 4 weeks of treatment.4,62
The course and prognosis of the acute disease in patients treated with diethylcarbamazine are generally benign, and 3 weeks of diethylcarbamazine therapy is curative in most patients. However, acute relapses related to reinfection or release of microfilaria from existing adult worms do occur in up to 20% of patients.4 Persons whose α1-antitrypsin levels remain low after initial treatment may be at greater risk of relapse.4,66 Patients who experience acute relapses often respond to additional treatment with diethylcarbamazine at higher doses of 2 to 4 mg/kg three times a day for 21 to 30 days. Alternatively, mild, chronic inflammation may persist, causing chronic interstitial lung disease, with persistent respiratory symptoms, radiographic findings, and hematologic and serologic abnormalities.62 Persistent clinical symptoms have been reported over 2- to 5-year follow-up periods in up to 13% of patients with TPE treated with a standard course of therapy. BAL in these patients reveals a mild, persistent eosinophilia. Persons with symptoms of longer duration are less likely to have a favorable treatment response. Alternative antifilarial drugs (e.g., ivermectin) or a trial of corticosteroids may be useful therapies for the chronic variant of the disease,4,62 although controlled studies of these agents are lacking. A subset of patients with apparent TPE may fail to respond to diethylcarbamazine; whether these patients have diethylcarbamazine-resistant TPE or disease due to other parasites is unclear, as current serologic testing does not distinguish between human lymphatic filarial antigens and antigens on certain other parasites.
Untreated disease usually persists for weeks to months. Untreated TPE may remit spontaneously, but it commonly recurs within months to years. It is important to treat TPE early in the course of the disease, since although seldom fatal, untreated TPE often leads to the development of irreversible pulmonary fibrosis.63,71
CHRONIC EOSINOPHILIC PNEUMONIA
CEP was first described as a clinical entity by Carrington et al.72 in 1969. Although CEP may develop in people of any age, the peak incidence occurs in persons 30 to 45 years of age.1,73,74 Women are affected approximately twice as often as men, and CEP has been reported during pregnancy and following radiation therapy for breast cancer.75 The female predominance is less obvious among patients whose disease begins after the age of 60. Most cases occur in Caucasians. Up to two-thirds have adult-onset asthma preceding (by several weeks to years) or arising concurrently with the occurrence of CEP.1,74,76,77 The asthma is often severe and may lead to fixed airflow obstruction (approximately 10% of patients) despite medical therapy.76 Most patients with CEP are nonsmokers. In addition, approximately one-third to one-half of patients have antecedent atopy, allergic rhinitis, nasal polyps, or urticaria.1
In contrast to idiopathic AEP, CEP typically has a subacute presentation, with symptoms present for weeks to several months before diagnosis.1,74 Common presenting complaints include dyspnea, low-grade fevers, malaise, drenching night sweats, and moderate (10 to 50 lb) weight loss.74,77 Cough, often dry initially and later productive of small amounts of mucoid sputum, is a virtually universal finding.74 Rhinitis or sinusitis may be present. Two of the nine patients described in Carrington’s original series had minor hemoptysis. Patients ultimately develop progressive dyspnea, which may be associated with wheezing in those with adult-onset asthma. Very rarely, patients with CEP may also have severe acute respiratory failure or ARDS, with severe hypoxemia requiring mechanical ventilation. There are no major extrapulmonary manifestations of CEP. Rarely, arthralgias, skin rash, diarrhea or colitis, mononeuritis, hepatitis, pericarditis or unexplained heart failure have been described, raising suspicion that there may be a continuum between CEP and EGPA.74 Indeed, cases have been reported wherein patients initially diagnosed with CEP later developed EGPA.78,79
Patients with CEP frequently manifest a moderate leukocytosis. The majority (66%–95%) have peripheral blood eosinophilia (usually >1000/mm3),74 with eosinophils constituting more than 6%, and typically up to 20% to 30% of their leukocyte differential.73,80 Leukocyte differentials with up to 90% eosinophils have been noted in this disorder. However, a lack of peripheral blood eosinophilia does not rule out the diagnosis, since eosinophilia may be absent in 10% to 30% of cases.73,77 Normochromic, normocytic anemia and thrombocytosis may be present. The C-reactive protein levels and the ESR are typically elevated (greater than 20 mm per hour),74 and IgE levels are elevated in up to one-half of cases. Analysis of BAL fluid reveals increased eosinophils, typically accounting for 40% or more of the white blood cell (WBC) differential, with a range from 12% to 95% reported.1,74,77,81 Urinary eosinophil-derived neurotoxin levels are also elevated.82 Blood and sputum cultures routinely fail to identify an infectious etiology in these patients.
The severity of pulmonary function abnormalities depends on the stage and severity of the disease. In the initial stage prior to treatment with corticosteroids, testing may reveal restrictive, obstructive, or normal physiology.74 Obstructive ventilatory defects, while more common in patients with a history of asthma, are also encountered in patients without pre-existing asthma. Restrictive physiology may result from changes in lung compliance due to acute eosinophilic infiltration of lung parenchyma. Diffusing capacity may be reduced and the alveolar–arterial oxygen gradient may be mildly elevated.74,77
In the original series, Carrington et al. described three radiographic features that are characteristic for CEP: (1) peripherally based, progressive dense infiltrates; (2) rapid resolution of infiltrates following corticosteroid treatment, with recurrences in identical locations; and (3) the appearance of infiltrates as the “photographic negative of pulmonary edema.”72,83,84 The pulmonary infiltrates associated with CEP are typically dense and patchy areas of airspace consolidation with ill-defined margins usually affecting the outer two-thirds of the lung fields (Fig. 71-3). Infiltrates are most commonly bilateral, tend to be located in the mid to upper lung zones, and may mimic loculated pleural fluid. They are frequently nonsegmental, subsegmental, or lobar in distribution. The characteristic “photographic negative of pulmonary edema” appearance (which occurs in <50% of cases) results if extensive infiltrates surround major portions of or the entire lung. Pleural effusions and cavitation are rare.73,85,86 The infiltrates may be migratory in up to 25% of cases. Occasionally, the chest radiograph can be normal.
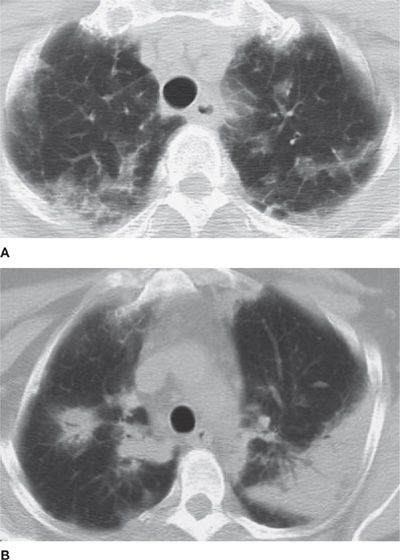
Figure 71-3 Radiographic appearance of chronic eosinophilic pneumonia (CEP). Variable computed tomography appearance of infiltrates in two patients with chronic eosinophilic pneumonia. Peripheral upper lobe–predominant infiltrates may have a ground-glass appearance (A) or may appear as regions of dense consolidation or nodular opacity (B).
Common CT scan findings include ground-glass opacities and areas of consolidation involving the middle and/or upper lung zones in peripheral regions of the lung.73,87–90 In addition, apparent unilateral or isolated lower lung zone involvement noted on chest radiography may prove to be bilateral and diffuse on CT scanning. Mediastinal adenopathy, which may be evident on conventional chest radiograph, may also be identified on CT scan.90 Less typical radiographic findings include nodular infiltrates, linear oblique or vertical densities, bronchial wall thickening, pleural effusion, and areas of fibrosis unassociated with anatomic divisions. Findings on CT scan may vary depending on the timing of the CT relative to the onset of symptoms. Typical areas of dense, peripherally located airspace consolidation are found in most cases within the first several weeks of disease onset. Streaky bandlike opacities may appear when symptoms have been present for more than 2 months.
The pulmonary lesions of CEP are characterized histopathologically by varying degrees of leukocytic infiltration of the alveolar airspaces and alveolar septae. These infiltrates are predominantly eosinophilic,49,72,77 with some associated macrophages, a small-to-moderate number of lymphocytes, occasional plasma cells, multinucleated giant cells, and an associated fibrinous exudate. Unlike AEP, the basal lamina may be disrupted49 but frank alveolar necrosis is absent. Eosinophilic microabscesses can be found. Focal edema of the capillary endothelium, focal type II epithelial cell hyperplasia, proteinaceous and fibrinous alveolar exudates can also be appreciated. Histologic evidence of proliferative bronchiolitis obliterans or bronchiolitis obliterans–organizing pneumonia may occur in up to one-third of cases, and a mild, nonnecrotizing microangiitis affecting predominantly the small venules may be seen. Biopsy specimens of lymph nodes from patients with intrathoracic lymphadenopathy reveal lymphoid hyperplasia and eosinophil infiltration.
The cause of CEP is unknown. No specific genetic predisposition for the disease has been identified, although CEP has been reported in identical twins, raising the question of a familial tendency toward the disease. An association has been reported between rheumatoid arthritis and CEP91 but no clear causal relationship has been identified.92 Although the precise immunopathogenesis of CEP is unknown, evidence suggests that Th2 helper T cells likely have a role in disease pathogenesis. Levels of the cytokines TARC-CCL17 and macrophage-derived cytokine 22 (MDC-22) and macrophage inflammatory protein 1-beta (MIP-1β/CCL14), which help recruit Th2 T cells are increased in CEP.74,84 Increased levels of the T-cell–derived eosinophil chemoattractant cytokines IL-5, eotaxin, and RANTES (CCL-5) are also elevated in BAL fluid of patients with CEP.74 Thus Th2 cells likely recruit and attract eosinophils to the lung.84 The number of regulatory (CD4+CD25+) T-cells is also increased in peripheral blood and BAL in CEP.93 The potential role of blood and lung tissue lymphocytes in the pathogenesis of CEP requires further study.
Several lines of evidence suggest that eosinophils also play a primary pathogenetic role in the pulmonary tissue damage seen in this disorder. Increased numbers of eosinophils appear in the peripheral blood and bone marrow before the onset of clinical disease, and eosinophilia is the predominant abnormality in BAL fluid. These eosinophils appear to be activated, since they show evidence of degranulation on electron microscopy,94 eosinophil-derived granule proteins (EDGPs) have been identified microscopically within the pulmonary parenchyma and microvasculature, increased concentrations of EDGP are identified in BAL fluid from patients with CEP compared to controls, and BAL-derived eosinophils express activation markers including class II major histocompatibility (MHC) antigens.95 Also, eosinophil-derived neurotoxin82 and leukotriene E496 are identified in the urine of patients with CEP, and inducible nitric oxide synthase (iNOS) is expressed on lung eosinophils.97 The processes that regulate eosinophil activation and degranulation in CEP are not clear. Evidence showing that class II MHC and other activation markers are expressed by BAL- but not blood-derived eosinophils suggests the presence of an immune inflammatory response compartmentalized within the lung. Data also suggest that eosinophils from the BAL fluid are more resistant to apoptosis than peripheral blood eosinophils in subjects with CEP.98
Of interest are the findings that immunoglobulins can augment eosinophil chemotaxis and degranulation in vitro, and that circulating immune complexes and elevated titers of IgE are noted in the context of clinical flares of the disease. To date, however, no clear causal relationship has been established between immunoglobulins and eosinophil activation in CEP. An association between CEP and diffuse pulmonary neuroendocrine cell hyperplasia has also been reported.99
The diagnosis of CEP is based on the presence of compatible clinical, radiographic, and BAL findings, and on the inability to document pulmonary or systemic infection or other known causes of eosinophilic lung disease. The clinical signs and symptoms of CEP are nonspecific, however, and blood eosinophilia and typical radiographic features may be absent in some cases. In most reported series, open lung biopsy has been required only rarely to establish the diagnosis. Transbronchial biopsy, usually performed to rule out other diagnostic entities, may reveal eosinophil and mononuclear cell infiltrates. Because of the rapid and dramatic responsiveness of CEP to steroid treatment, a therapeutic trial of steroids is often useful in establishing the diagnosis. Failure to document rapid clinical improvement should alert the clinician to consider other diagnoses. The differential diagnosis of CEP includes drug-induced eosinophilic pneumonia, infection (especially TB, fungal diseases such as cryptococcosis and parasitic disease), sarcoidosis, Loeffler syndrome, desquamative interstitial pneumonitis, cryptogenic organizing pneumonia, ABPA, chronic hypersensitivity pneumonitis, acute idiopathic eosinophilic pneumonia, EGPA, and eosinophilic granuloma.
CEP rarely resolves without therapy and if left untreated may result in pulmonary fibrosis.100 Corticosteroids are the mainstay of therapy for CEP. Dramatic clinical, radiographic, and physiologic improvements have been documented following steroid treatment in all series reported.1,74,87 Even patients presenting with severe respiratory failure may respond well to steroid treatment. In most cases, treatment with steroids leads to defervescence within 6 hours, reduced dyspnea, cough, and blood eosinophilia within 24 to 48 hours, resolution of hypoxia in 2 to 3 days, radiographic improvement within 1 to 2 weeks, complete resolution of symptoms within 2 to 3 weeks, and normalization of the chest radiograph within 2 months.1,73,77 No comparative studies exist to determine optimum treatment doses or duration of steroids, but one recommended regimen is prednisone 0.5 mg/kg/d (40–60 mg a day) continued until 2 weeks after resolution of symptoms and radiographic abnormalities, generally for 4 to 6 weeks. The dose of prednisone can then be tapered slowly by 0.25 mg/kg/d and then continued for the subsequent 8 weeks. Treatment is usually maintained for at least 3 months and optimally for 6 to 9 months; during this phase prednisone dosing can be decreased by 5 mg every 4 weeks. Shorter courses of prednisone may also be effective.74
The prognosis of CEP is generally favorable.101 In steroid-treated patients, morbidity and mortality directly related to CEP are low. Patients may require 1 to 3 years of initial steroid treatment to control the disease,1 and up to 50% may require long-term maintenance treatment (2.5–10 mg prednisone a day) to remain disease-free.74 The lowest possible dose of steroid that suppresses disease activity should be used. Some patients may respond to high doses (e.g., 1000–1500 μg/24 h) of inhaled corticosteroids, allowing discontinuation of oral steroids, although inhaled steroids alone as initial therapy are inadequate.
Clinical, hematologic, or radiographic evidence of relapses are common, occurring in 50% to 80% of cases when steroids are tapered or discontinued.1,73,74,77 Relapses may involve radiographic infiltrates in the same or different anatomic distribution compared to the original disease. Relapsing CEP must be distinguished from the development of new or worsening asthma. No obvious factors exist to identify persons who are likely to relapse or require long-term steroids, although relapses are more common in persons treated initially with a short course (<6 months) of steroids. Regular long-term treatment of patients with CEP and asthma with inhaled corticosteroids may reduce the risk of CEP relapses.76 Multiple recurrences may occur in anyone. Relapses should be managed by increasing the prednisone dose to ≥40 mg per day until 2 weeks after symptom control has been achieved, with gradual taper thereafter. The reinstitution of steroids generally leads to improvement, and relapses do not appear to indicate a worse prognosis, increased likelihood of treatment failure, or increased morbidity. The anti–IL-5 monoclonal antibody omalizumab has also been used successfully as a steroid-sparing agent in the treatment of CEP.102
ALLERGIC BRONCHOPULMONARY ASPERGILLOSIS (MYCOSIS)
ABPA is a disorder caused by a complex hypersensitivity response to inhaled fungal antigens.103–110 Since the disease is most commonly induced by Aspergillus species, it is usually known as ABPA. When induced by non-Aspergillus species, the syndrome is called allergic bronchopulmonary mycosis. A comprehensive discussion of ABPA is provided in Chapter 48. Highlights of the disorder are discussed here.
ABPA occurs most commonly in immunocompetent patients and complicates 1% to 2% of cases of persistent asthma and 7% to 14% of cases of chronic steroid-dependent asthma,111 most often among patients in their third to fourth decade. It also complicates up to 15% of patients with cystic fibrosis (CF), most often during the teen years.105,112 Rare cases lacking a history of asthma but meeting the other major diagnostic criteria (summarized in Table 71-5) have been reported.113 The diagnosis of ABPA is based on appropriate clinical features in combination with supporting serologic and radiologic findings.104



