Chapter 44 Sustained Ventricular Tachycardia with Heart Disease
The definition of sustained VT has undergone evolution. Although no formal definition has been adopted for spontaneous sustained VT compared with VT induced by programmed stimulation, two versions are in common use. In the Electrophysiologic Study Versus Electrocardiographic Monitoring (ESVEM) trial, a 15-second duration was considered sustained VT.1 Alternatively, many physicians prefer to use 30-second duration or evidence of hemodynamic compromise warranting earlier termination to define VT, as in the case of induced VT. This chapter provides an overview of the epidemiology, pathology, electrophysiology, and management of sustained VT in patients with structural heart disease. In such patients, VT can be conveniently divided into (1) VT caused by, or related to, a “fixed arrhythmogenic substrate” and (2) VT in which the substrate is present intermittently during transient abnormalities in cardiac electrophysiological structure and function (e.g., with changes in the neurohormonal milieu).
Etiology and Pathologic Anatomy
In general, the majority of hearts in patients with a previous history of VT reveal varying types of pathologic changes, with areas of healthy myocardium interspersed among the pathologic processes.2–4 The most common cause of VT is coronary artery disease (CAD), in either its acute phase or chronic phase. During the acute phase, VT is believed to be caused by altered physiologic, biochemical, or metabolic states. In the chronic phase, a re-entry circuit is created at the junctional areas of the healthy myocardium and scar tissue. The infarct zone is surrounded by varying degrees of border zone with surrounding but damaged myocardial fibers. In general, the infarct size is quite large in these patients and may be related to the development of a ventricular aneurysm.2–4
VT also frequently occurs in cardiomyopathy. Any disease state that affects the heart in a chronic process can eventually lead to a cardiomyopathy. Thus VT is expected to occur in advanced cardiomyopathy during the end stage of heart diseases such as CAD, myocarditis, valvular disease, hypertensive heart disease, familial types of cardiomyopathy, and in many other disease states. This cardiomyopathy can have several etiologies; however, the most common type is an idiopathic form. In some patients, “idiopathic” cardiomyopathy may be the end result of previous viral myocarditis. Pathologically, in most cases of cardiomyopathy, the ventricular myocardial cells show varying types of histologic features such as hypertrophy of cells and varying stages of degenerative changes in the cells and fibrosis, with or without the presence of chronic inflammatory cells. Varying degrees of myocardial fiber damage can provide a substrate for a re-entry circuit at the junctional areas with the healthy myocardium.3
Myocarditis of any type, in the acute phase or the chronic phase, may cause VT that may result in SCD. The heart may be normal at the gross anatomic level, but microscopic examination may reveal an interstitial type of a myocarditis in the ventricular myocardium, including the bundle branches. In many patients, clinical history may be essentially unremarkable, or a history of a mild attack of influenza several weeks before death may be present.2–4
Fibrotic scars in the ventricular myocardium are often seen in young victims of SCD who otherwise have a normal heart. They may be associated with pathologic changes in the peripheral conduction system such as the branching atrioventricular (AV) bundle and bundle branches. The focal scars in the ventricular myocardium, surrounded by healthy myocardium, may form an anatomic substrate for ventricular arrhythmias that promotes re-entry or abnormal automaticity. The etiology of the fibrotic scars in the ventricular myocardium and the beginning of the bundle branches currently is unknown. One hypothesis is that these may represent the end result of an autoimmune reaction or an allergic state that may or may not be related to a silent form of a previous myocarditis.2–4
Uncommon Types of Cardiomyopathy
Less commonly, VT is seen in mitral valve prolapse. Several pathologic abnormalities in cases of mitral valve prolapse are associated with VT.2–5 A variety of pathologic findings such as a right-sided AV bundle, fibrotic scars in the ventricular septum, degenerative changes in the conduction system, and arteriolosclerosis, may be seen in these patients.
In arrhythmogenic right ventricular dysplasia, the anterior wall of the right ventricle can be partially or completely replaced by fibro-fatty tissue. Some intact myocardial cells may be scattered within the fatty tissue. Similar findings extend to the ventricular septum and the left ventricular myocardium. This often is associated with necrosis of cells and mononuclear cell infiltration. Varying amounts of degenerative changes in the myocardium are present in the right and left ventricles. A segmented and looping left-sided AV bundle has been reported. Small-vessel disease of the ventricular septum may also be present. Thus, in addition to acquired pathologic changes, congenital abnormalities of the conduction system in the sinoatrial, AV node, and AV bundle also occur.2–46
In hypertrophic cardiomyopathy, the heart is hypertrophied and enlarged. Hypertrophy of the interventricular septum is seen in varying degrees. The AV node may be partly or mostly situated within the central fibrous body and occasionally partly embedded in the tricuspid valve annulus or at the aortic-mitral annulus. The AV nodal artery is usually thickened and narrowed. The sinoatrial (SA) and AV nodes frequently are infiltrated with fat. The AV bundle may be on the right side of the ventricular septum with loop formation and fibrosis of the branching bundle. Focal, fibrotic scars in the ventricular myocardium are associated with myocardial fiber disarray and arteriolosclerosis in the summit of the ventricular septum.2–47
Infiltrative Diseases of the Myocardium
VT may occur in sarcoidosis and amyloidosis. An infiltrative disease of the myocardium such as sarcoidosis may affect the branching AV bundle and the bundle branches as well as the ventricular myocardium. Likewise, amyloidosis, in either the primary or the secondary form, may involve the ventricular myocardium, including the bundle branches. In 70% of cases of primary amyloidosis, the heart is involved, often with disruption to the entire conduction system. The infiltration of amyloid in the heart eventually results in a restrictive cardiomyopathy, and SCD is commonly seen.2–48 The heart is affected in approximately 13% to 25% of cases of sarcoidosis. Cardiac involvement often is associated with lymph node and lung involvement. Sarcoidosis has a predilection to affect the posterior wall of the left ventricle with aneurysm formation. Sarcoid granulomas may be present in the conduction system and the surrounding myocardium (Figure 44-1). In general, infiltrative lesions can cause conduction disturbances and provide a substrate for re-entrant or automatic VT.2–48
Primary Electrical Diseases
Supraventricular arrhythmias are well known to occur in pre-excitation syndrome. However, ventricular arrhythmias also can occur in this entity. Pathologically, the accessory pathway is obvious on either the left or right side, associated with cardiomyopathy and fibroelastosis of the left ventricle, along with hypertrophy and degenerative changes of the myocardium.2–49
Familial Q-T interval prolongation is predominantly an autosomal dominant disorder associated with syncopal episodes, VT, and SCD. Recently, abnormal genes have been identified on several chromosomes in patients with congenital prolonged Q-T interval. Genetic heterogeneity has been documented in linkage studies, in which loci on several chromosomes have been identified. Furthermore, mutations in ion channel genes on chromosomes 3, 7, and others have been identified as related to other forms of long QT syndrome.2–410 Pathologically, there are marked fatty infiltration in the approaches to the AV node; a lobulated AV bundle with or without loop formation; arteriolosclerosis; and focal fibrosis of the summit of the ventricular septum seen predominantly on the right side. In addition, fibrotic changes are seen to a varying degree in the AV bundle and bundle branches with chronic inflammatory cells in the ventricular myocardium. Of note, pathologic findings do exist in congenital long QT syndrome leading to SCD.2–410
Skeletal Muscle Disorders
The conduction system frequently is affected in congenital myotonic dystrophy. This progressive, generalized disease is characterized by typical atrophy of skeletal muscles with associated myotonia. Various types of arrhythmias are known to occur in this disease. Pathologically, degenerative changes in the smooth muscles of the cardiac vessels in the left atrium and the aorta with fatty infiltration in the approaches to the AV node, fibrosis of the bundle branches, summit of the ventricular septum, and varying degenerative changes in the myocardium are present. These could form a substrate for AV block and VT.3,11
Kearns-Sayre syndrome is characterized by progressive external ophthalmoplegia, retinitis pigmentosa, and AV block rather than VT. Progressive, degenerative changes affect the entire heart. The conduction system is replaced by fibrotic destruction of the bundle branches with fibro-fatty replacement. Some muscle cells may reveal hypertrophy; others become atrophied with perineural and perivascular fibrosis, eventually leading to cardiomyopathy. Although AV block frequently occurs in this disease, electrophysiological studies have demonstrated that the disease affects the entire His-Purkinje system. These findings in the conduction system and ventricular myocardium may form an anatomic basis for VT.3,12
Tumors and Parasitic Diseases of the Heart
Any tumor, either primary or secondary, can produce ventricular arrhythmias, as can parasitic infiltration. Hydatid cyst infiltration can result in fibrosis in the perimeter tissues around the cyst. This can permit re-entry and may result in VT.3,13
Aneurysm and Diverticulum of the Heart
Aneurysm and diverticulum of the heart may occur in either the right or left ventricle of an otherwise normal heart, and VT may be the first clinical manifestation.3,13 Pathologic anatomy reveals a large, wide-mouthed aneurysm, but its microscopic appearance can vary with the etiology.
Ventricular Tachycardia in Postoperative Patients with Congenital Heart Disease
Ventricular arrhythmias are known to occur in postoperative patients with congenital heart diseases such as tetralogy of Fallot, aortic stenosis, or other congenital cardiac anomalies. VT can appear many years after the surgery. Fibrotic scars along with healthy myocardium are seen, for example, at the outflow tract of the right ventricle or in any other area in the heart. This substrate can result in re-entry circuits leading to VT.2–4,14,15 In aortic stenosis, cardiomegaly may occur with myocardial fibrosis.16
Iatrogenic Disorders
Antiarrhythmic drugs may alter the physiological, metabolic, and biochemical states of the myocardium, the conduction system, or both and give rise to VT, which may or may not be transient. Likewise, the various resuscitative techniques used and catheter ablation of the myocardium may alter myocardial tissue, leading to formation of fibro-fatty scar and chronic inflammation. This may become a future substrate for VT.2–4,13,17
Familial Ventricular Tachycardia
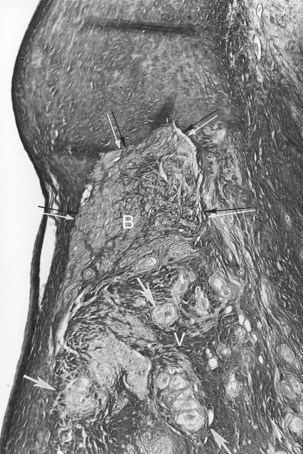
Pathologically, the conduction system and the ventricular myocardium may show degenerative changes with mononuclear cell infiltration and fat to varying degrees. In other individuals, atrophy of the branching part of the AV bundle, with almost complete absence of both right and left bundle branches, may be present. These findings suggest a genetic abnormality of the conduction system that leads to degenerative changes, inflammatory phenomena, and susceptibility to ventricular arrhythmias that cause SCD.18,19 The anatomic substrate may originate from the myocardial disarray that can be present in either the ventricular septal myocardium or the conduction system. Left or right ventricular septal hypertrophy may also cause degeneration of the AV node, AV bundle, and the main left bundle branch (Figure 44-2).3,4,20
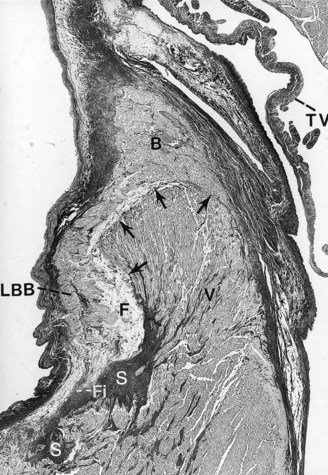
 -year-old boy with a history of exercise-related syncope died suddenly while swimming. He had a family history of sudden cardiac death involving three consecutive generations, including a brother. The electrocardiogram and cardiac catheterization results were normal. During electrophysiological studies, with extrastimulus testing, he had polymorphic nonsustained ventricular tachycardia; during stage 5 of the Bruce protocol, he had a run of nonsustained ventricular tachycardia. Photomicrograph of the branching atrioventricular bundle being compressed by the right ventricular septal muscle at the region of the posterior radiation of left bundle branch (Weigert-van Gieson stain ×22.5). B, Branching bundle; F, fatty metamorphosis; Fi, fibrosis and linear change of left bundle branch; LBB, posterior radiation of left bundle branch; S, increased sclerosis on the mid-septal area on the left; V, summit of the ventricular septum. Arrows point to the pressure of the right ventricular septal hypertrophy on the atrioventricular bundle at the level of the posterior radiation of left bundle branch.
-year-old boy with a history of exercise-related syncope died suddenly while swimming. He had a family history of sudden cardiac death involving three consecutive generations, including a brother. The electrocardiogram and cardiac catheterization results were normal. During electrophysiological studies, with extrastimulus testing, he had polymorphic nonsustained ventricular tachycardia; during stage 5 of the Bruce protocol, he had a run of nonsustained ventricular tachycardia. Photomicrograph of the branching atrioventricular bundle being compressed by the right ventricular septal muscle at the region of the posterior radiation of left bundle branch (Weigert-van Gieson stain ×22.5). B, Branching bundle; F, fatty metamorphosis; Fi, fibrosis and linear change of left bundle branch; LBB, posterior radiation of left bundle branch; S, increased sclerosis on the mid-septal area on the left; V, summit of the ventricular septum. Arrows point to the pressure of the right ventricular septal hypertrophy on the atrioventricular bundle at the level of the posterior radiation of left bundle branch.Changes on the Right Side of the Ventricular Septum Related to Premature Aging
Premature aging occurs in the summit of the ventricular septum in some individuals, with degenerative changes of the branching bundle and the right bundle branch. These changes are usually associated with arteriolosclerosis of the summit of the ventricular septum and often are seen in SCD in teenagers.3,4,21
Idiopathic Ventricular Tachycardia Caused by Congenital Abnormalities of the Conduction System
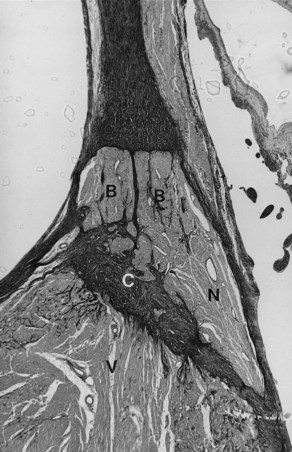
Chronic recurrent right VT with QRS morphology of a left bundle branch block (LBBB) pattern in a normal heart, normal coronary arteries, and normal cardiac catheterization findings has been well documented, especially in young patients. However, an anatomic substrate for this abnormality has only been occasionally reported.22 In one report, a 13-year-old boy with a history of recurrent VT died suddenly. A right-sided, markedly septated AV bundle was found at autopsy. The AV node formed the AV bundle, and the node-bundle junction was ill defined (Figure 44-3). The penetrating AV bundle cells were not well defined as typical cells of the AV bundle, and the latter remained on the right side and eventually became the right bundle branch. In addition, patchy fibrotic scars were present in the right ventricular myocardium. A right-sided, markedly septated, undifferentiated AV bundle that eventually continues as the right bundle branch has been hypothesized to cause recurrent right VT in an otherwise normal heart.2,3,22
Basic Electrophysiology
Ischemia and Ventricular Tachyarrhythmias
The ventricular arrhythmias caused by myocardial ischemia and infarction occur in several distinct phases. A ventricular arrhythmia can be induced by acute ischemia and reperfusion. It usually occurs between 2 and 30 minutes after acute coronary artery occlusion, when the changes caused by ischemia are still reversible. Arrhythmias associated with the development of myocardial infarction can be categorized as delayed arrhythmias, usually occurring between 5 and 48 hours, late arrhythmias occurring after days to weeks, and chronic arrhythmias occurring months to years later.23,24 Delayed arrhythmias such as slow VT and accelerated idioventricular rhythms rarely degenerate into VF and are caused by abnormal automaticity of Purkinje fibers overlying the infarct.
Data obtained in 4- to 5-day-old canine infarcts show that the healing infarct undergoes structural and functional changes. The surviving epicardial cells overlying the infarct have abnormal action potentials with diminished upstrokes with loss of the plateau and shorter action potential duration. The density and kinetics of a number of ion channels are altered, and sodium ion and calcium ion currents are reduced, as are transient outward potassium ion currents and the delayed and inward rectifying potassium ion currents.25 During this stage, re-entrant VT in the so-called epicardial border zone (the layer of surviving cells overlying the infarct) can easily be induced by premature stimuli. Both the cellular abnormalities, as well as a re-distribution of intercellular gap junctions, play a role in determining the “substrate” for re-entry.26 These VTs may degenerate into VF, especially in the presence of a high sympathetic tone, but this is uncommon.24
In the following few weeks, transmembrane action potentials of the surviving cells gradually return to normal as most of the ion channels recover; by 2 months, action potential configuration in both canine and human infarcts is completely normal (Figure 44-4).27,28 When the healing phase of myocardial infarction is over and when the fully healed phase begins are difficult points to ascertain. The electrophysiological substrate for VT likely develops gradually over several weeks and remains stable from several months to 15 to 20 years.29
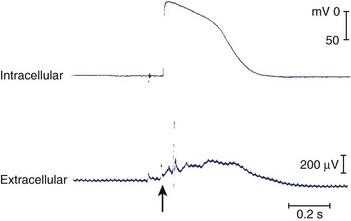
The Substrate for Re-entrant Tachycardia in the Human Heart with a Healed Infarct
In the prethrombolytic era, the incidence of sustained monomorphic VT after discharge from the hospital in patients surviving a myocardial infarction (MI) has been reported to be approximately 3% and that of nonsustained VT as 10% to 20%.30 Although thrombolysis has drastically reduced arrhythmic events early after MI, with an incidence of sustained VT lower than 1%, post-infarction arrhythmias have certainly not become irrelevant.31 With improved therapy in the acute stage of MI, more patients survive, but many survivors have left ventricular dysfunction. Ventricular dysfunction is known to be the most important risk factor for SCD, as verified in the European Myocardial Infarct Amiodarone Trial (EMIAT). The combination of arrhythmic death and resuscitated cardiac arrest occurred in 8.6% of patients.32 How many of these patients developed sustained VT that degenerated into VF is unknown, as is how many had an episode of acute ischemia that induced the lethal arrhythmia. Still, despite intensive therapy, arrhythmic events in patients who have had MI, especially in the presence of ventricular dysfunction, remain an important cause of death.
Reconstruction of the re-entrant circuits and the mechanism of slow conduction in the human heart with a healed infarct is based on observations made during mapping-guided surgery in patients with infarct-related VT as well as on studies on isolated, Langendorff-perfused human hearts and isolated papillary muscles from patients with infarcts undergoing cardiac transplantation.28,33–35 Figure 44-5 shows the activation map of one beat of a VT induced by programmed electrical stimulation in an isolated, Langendorff-perfused heart. The endocardial surface of the left ventricle is schematically depicted with the left ventricle folded out by making a vertical cut along the left anterior descending artery. The black zone indicates the infarcted area.
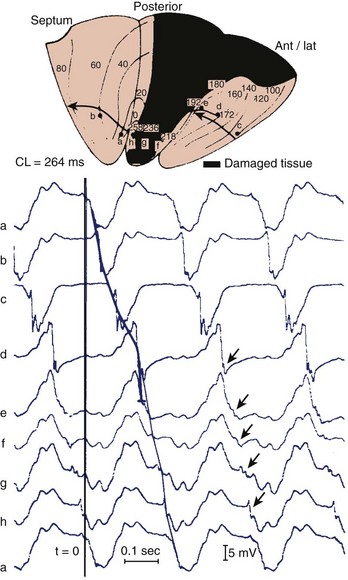
Selected extracellular electrograms, recorded simultaneously from the endocardium by a balloon electrode inserted into the ventricular cavity, are shown in Figure 44-5 as well. The cycle length of this tachycardia was 264 ms. The site of earliest activation during this tachycardia was in a small area near the apex, on the border of the septum and the posterior wall, at the margin of the infarcted region (the area encircled by the 0-ms isochrone). Activation spread from this area to the left in the figure (from a to b) and continued to the anterior wall (from b to c to d to e) to reach the other margin of the infarct after 192 ms (near site e). Although activation at this margin seems to have died out, because of the slow positive deflection recorded at site e, the presence of small deflections indicated by the arrows over the infarcted zone suggests that the spread of activation continued via tracts of surviving muscle within the infarct, through sites f, g, and h, to reach the opposite side of the infarct after 258 ms, re-exciting the area of the 0-ms isochrone and completing a large re-entrant circuit around the circumference of the ventricle.
Subsequent histologic studies of this region confirmed the presence of surviving myocardial muscle bundles, several millimeters below the endocardial surface that connected both margins of the infarct, completing the re-entrant circuit. Figure 44-6 shows superimposed drawings of many histologic sections of this region, where viable muscle is in black and connective tissue in white. Similar tracts were found in other hearts, embedded within the scar of the healed infarct. Such tracts could be localized in the subendocardium, the subepicardium, and intramurally. The myocardial fibers were sometimes arranged in a parallel fashion along the long axis of the tract, allowing relatively rapid transmission of the re-entrant impulse from one side of the infarct to the other. However, in other hearts the fibers were oriented transverse to the direction of impulse propagation; in these cases, transmission through the infarct was very slow.

Figures 44-7 and 44-8 show the mechanism of such “slow” conduction. Figure 44-7 shows selected electrograms from an infarcted human papillary muscle to illustrate the highly fragmented waveforms, typical when connective tissue is intermingled with viable myocardium. In this muscle, the delay between the deflections at the extreme electrode terminals of the multiple electrode (distance, 1.4 mm) was 45 ms, which would correspond to an overall conduction velocity on the order of 3 cm/sec. As illustrated in Figure 44-8, this apparent slow conduction was caused by “zigzag” conduction in small muscle bundles separated by collagenous septa. Activity in the muscle tracts proceeded both toward the site of stimulation A and away from it. Many tracts were dead-end pathways. The actual pathway of activation is plotted in panel B. Although the shortest distance between A and B was 1.2 mm, the length of the zigzag pathway was 25.2 mm. In 10 papillary muscles, conduction velocity parallel to the fiber orientation was, on average, 79 cm/sec, which indicates that both the active electrical properties (action potential amplitude and upstroke velocity) and passive electrical properties (longitudinal coupling resistance) were normal. Conduction velocity at the bifurcation points was only 49 cm/sec, indicating that in addition to the pathway length, impedance mismatch at bifurcations also contributed to activation delay.
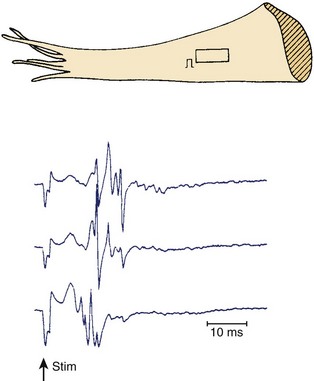
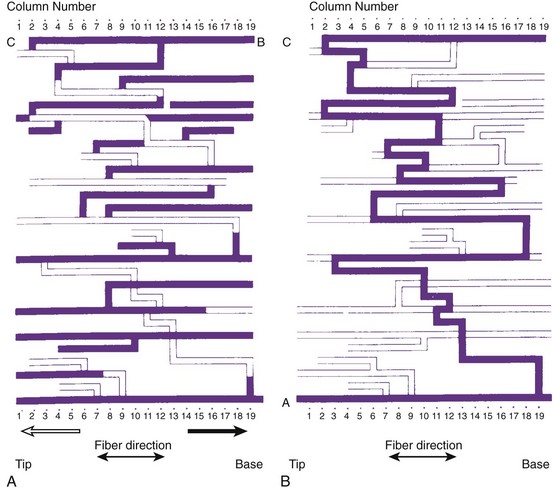
FIGURE 44-8 Map illustrating the spread of activation of the rectangular area shown in the inset of Figure 44-4. In the solid tracts of A, activation moves away from the site of stimulation (site A); in the open tracts, activation moves toward the site of stimulation. B, The tortuous route that activation followed to reach site B.
(Modified from De Bakker JMT, van Capelle FJL, Janse MJ, et al: Slow conduction in the infarcted human heart: “Zigzag” course of activation, Circulation 88:915–926, 1993.)
Thus, in the human heart with a healed infarct, the substrate for re-entry is formed by the surviving fibers within the infarct. In addition to a substrate, a trigger is needed to initiate the tachycardia. This trigger likely occurs in the myocardium remote from the infarct. In the presence of progressive left ventricular dysfunction, these myocardial zones can undergo arrhythmogenic remodeling. Furthermore, the properties of the noninfarcted myocardium can determine whether VT degenerates into VF. Dispersion of refractoriness in the noninfarcted myocardium is three times larger in patients who have had an MI and subsequently develop VF than in those in whom the VT remains monomorphic and hemodynamically stable.36
Role of Cardiac Remodeling in Ventricular Tachycardia
The heart may respond to a variety of abnormal environmental stimuli by altering gene expression or the functional properties of proteins. This ultimately leads to both functional and structural cardiac alterations that may be arrhythmogenic. These processes are referred to as cardiac remodeling. Common to all studies on the hypertrophic and failing ventricular myocardium is a prolonged action potential, especially at slow heart rates.37 This may be considered an adaptive process. In the setting of a prolonged action potential, the intracellular calcium transport is increased, causing enhanced force of myocardial contraction. It can be argued that a prolonged action potential may also protect the heart against re-entrant excitation. However, prolonging repolarization may be arrhythmogenic. Drugs, both cardiac and noncardiac, can prolong the action potential. A risk of development of early afterdepolarizations (EADs) and torsades de pointes (TdP) does exist in this situation.38 An EAD-induced premature beat generally causes re-entry only in the presence of a marked increase in dispersion of repolarization.38 In the hypertrophic and failing myocardium, increased dispersion in repolarization (or refractoriness) has been reported.36,39–41 In hearts with a healed infarct, repolarization of normal, lateral border and infarct-zone cells is nonuniform, with myocardial cells showing different degrees of action potential disturbance and prolongation. Many of these cells, especially those close to the infarct border, show post-repolarization refractoriness.25 Several reasons for the increased dispersion in repolarization exist: unequal distribution of remodeled ion channels, decreased expression of gap junctional connexins and an altered distribution of gap junctions with the development of fibrosis, and changes in autonomic innervation.41–46
Changes in ionic currents contributing to repolarization are seen. The most consistent finding is a reduction in the transient outward current, Ito.37,42,43 Although this current is very important in determining action potential duration in small mammals such as the rat, its downregulation probably does not have much effect on action potential duration in the hearts of large mammals. It does change the level of the plateau phase of the action potential, and it can affect other currents activated later during the action potential. Both rapid and slow components of the delayed rectifier, Ikr and Iks, are reduced in rabbits with pacing-induced heart failure.47 The inward rectifier current, IK1, can decrease, remain unchanged, or increase.37 If IK1 is reduced, it will lead to an unstable resting potential; when it is unregulated, pacemaker activity could result.48,49 The L-type calcium current is either unchanged or decreased, and it is unlikely that this current contributes to action potential prolongation.50 A role may exist for the late sodium current, which is increased.51 For most of these remodeled currents, data on regional differences are insufficient. However, in the presence of enhanced β-adrenergic activity, transmural dispersion in repolarization increases because of augmentation of residual Iks in the epicardial and endocardial layers, resulting in action potential shortening.52 But this does not occur in mid-mural M cells, in which Iks is intrinsically weak.52 A decrease in electrical cell-to-cell coupling, either by a decrease in expression of connexins or by the development of microscopic fibrosis in hypertrophied myocardium, reduces electronic current flow and will unmask intrinsic differences in action potential duration. The well-coupled myocardium will attenuate these differences.
Ischemia and infarction result in both afferent and efferent parasympathetic and sympathetic dysfunction in regions apical to the area of infarction. The denervated but otherwise normal myocardium develops adrenergic supersensitivity, and therefore the response to circulating catecholamines is exaggerated.45 The hypothesis of nerve sprouting in ventricular arrhythmias and SCD states that myocardial infarction results in nerve injury followed by sympathetic nerve sprouting and regional, heterogeneous myocardial hyperinnervation, which, together with electrical remodeling, leads to heterogeneous distribution of repolarization and ventricular arrhythmias.46
It is generally agreed that intracellular calcium handling is compromised in heart failure, but reports on virtually all components involved in calcium homeostasis are contradictory.37,53–55 It is generally accepted that adenosine triphosphate–dependent calcium accumulation by the sarcoplasmic reticulum is decreased. In the presence of a prolonged action potential and altered calcium homeostasis, both EAD and delayed afterdepolarization may occur.43,49 In hearts with an infarct, in which an anatomic re-entrant circuit may be present, either premature ventricular depolarizations caused by these afterdepolarizations or salvos of triggered activity may initiate sustained re-entrant tachycardias. In hypertrophied and failing hearts without an infarct, with fibrosis and increased dispersion of repolarization, these triggers may equally initiate re-entrant tachycardias. In animal models of heart failure (e.g., rabbits), combined volume and pressure overload resulted in nonsustained VT developing in more than 50% of animals, and SCD was common.56 However, it is not certain whether SCD is always caused by VT degenerating into VF. In humans with end-stage heart failure as well as in rabbit models of heart failure, bradycardia, asystole, and electromechanical dissociation have been documented to cause SCD.57–59
Clinical Presentation
The symptoms associated with sustained VT can range from an asymptomatic patient to cardiovascular collapse resulting in circulatory arrest and unconsciousness. Clinical literature dating to the early twentieth century has documented case reports of sustained VT without symptoms or minimal symptoms. Remarkably, some of these episodes may last weeks or even months. These patients may have only minimal or no palpitations, that, when present, can be regarded as an insignificant symptom. Figure 44-9 shows a patient with incessant VT who, in 1981, had palpitations with mild dyspnea during a single sustained slow VT episode that lasted 1 week, despite the presence of severe left ventricular systolic dysfunction. The most common symptoms of sustained VT are palpitations, dyspnea, angina, hypotension, and near or frank syncope. The severity of symptoms is related to several factors, including tachycardia rate, morphology, severity of left ventricular dysfunction, preload, and coexisting diseases such as CAD. The symptom complex often is defined by the hemodynamic impact of the arrhythmia. In an early study, Saksena and colleagues proved that VT resulted in impaired left ventricular relaxation and subsequently systolic dysfunction and decline in negative and positive dP/dt.60 Tachycardia rate and pre-existing LV dysfunction were important variables, as was preload. Faster VT rates and low preload predisposed to hypotension and syncope. Hamer noted similar associations with syncope during VT.61 In a study of defibrillator recipients, presyncope or syncope preceding delivery of implantable cardioverter defibrillator (ICD) therapy was determined by faster events in the “ventricular fibrillation” zone.62 However, the absence of hemodynamic compromise does not exclude the diagnosis of VT. An axiom holds that wide QRS tachycardia in the presence of history or electrocardiogram (ECG) evidence of myocardial infarction is due to VT until proven otherwise by subsequent investigation.
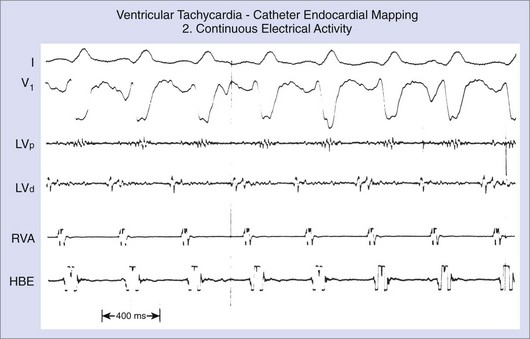
Electrocardiography
Electrocardiographic Diagnosis of Ventricular Tachycardia
Twelve-lead ECGs are particularly useful to differentiate supraventricular tachycardia (SVT) from VT. Distinguishing these arrhythmias is more difficult when aberrant intraventricular conduction occurs during SVT, but several criteria have been proposed to improve diagnostic accuracy.63–69 AV dissociation is the most specific ECG criterion for the diagnosis of VT in recordings of wide QRS complex tachyarrhythmias. Certain rare types of SVT (e.g., AV node re-entry with retrograde block, junctional tachycardia using a nodo-ventricular fiber with retrograde atrial block) may mimic this finding. AV dissociation is seen in only 21% of ECG recordings with VT and may be difficult to identify with absolute certainty.65 When AV dissociation is not apparent, other criteria that depend on whether the QRS resembles LBBB or RBBB must be used.
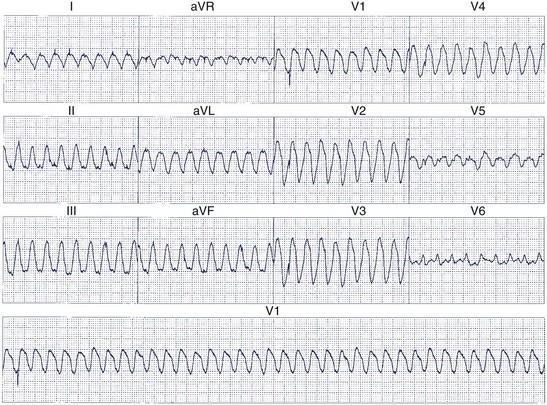
When the ventricles are structurally normal, QRS morphology is an excellent marker of the arrhythmia origin. VT with an LBBB configuration in lead V1 can have its origin in the right ventricle or the intraventricular septum (either right or left side of the septum) (Figure 44-10). A QRS frontal-plane axis directed inferiorly (dominant R waves in leads II, III, and aVF) indicates an origin in the cranial aspect of the heart (e.g., anterior wall of the left ventricle or the right ventricular outflow tract). A QRS frontal plane axis directed superiorly (dominant S waves in leads II, III, and aVF) indicates initial depolarization arising in the inferior wall of the left or right ventricle. Dominant R waves in leads V3 to V4 favor a location of the focus nearer the base of the heart than the apex. Dominant S waves in these leads favor a more apical location. VT with a typical LBBB or RBBB QRS configuration suggests bundle branch re-entry as a mechanism. When areas of ventricular scarring are present, the QRS morphology is less reliable and is sometimes quite misleading.
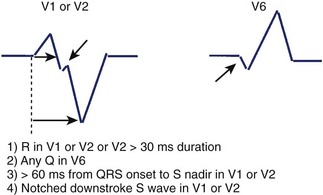
Kindwall observed several characteristic patterns found in patients with VT complexes resembling LBBB and evaluated four criteria to distinguish VT with LBBB pattern from SVT with aberrant conduction.66 All patients in the study had a predominantly negative QRS in V1 and a QRS duration greater than 120 ms. Four electrocardiographic criteria were evaluated in this study: (1) R wave in lead V1 or V2 of greater than 30-ms duration, (2) any Q wave in lead V6, (3) a duration of greater than 60 ms from the onset of the QRS to the nadir of S wave in lead V1 or V2, and (4) notching on the downstroke of the S wave in those leads. Table 44-1 summarizes the sensitivity and predictive accuracy of these criteria. None of the criteria, by itself, was very sensitive, but all patients with VT had at least one of these criteria. The specificity remained high (89%) when the combined criteria were used, and the predictive accuracy was excellent (Figure 44-11). Left-axis deviation was of no value in distinguishing VT from SVT in this study. These criteria are easily measured and provide a practical approach to differentiating VT from SVT.
Table 44-1 Sensitivity and Predictive Accuracy of Electrocardiographic Criteria to Distinguish VT Resembling LBBB from SVT with LBBB Aberration
| PREDICTIVE CRITERIA* | SENSITIVITY (%) | ACCURACY (%) |
|---|---|---|
| R > 30 ms in lead V1 or V2 | 36 | 100 |
| Any Q in V6 | 55 | 98 |
| Duration >60 ms from QRS to S nadir in lead V1 or V2 | 63 | 98 |
| Notched downstroke S wave | 36 | 97 |
| Any of the above present | 100 | 96 |
LBBB, Left bundle branch block; SVT, supraventricular tachycardia; VT, ventricular tachycardia.
* Diagnostic criteria for VT with a morphology resembling left bundle branch block.
From Kindwall KE, Brown J, Josephson ME: Electrocardiographic criteria for ventricular tachycardia in wide complex left bundle branch block morphology tachycardias, Am J Cardiol 61:1279–1283, 1988.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


