CHAPTER 96 Surgical Management of Hypertrophic Cardiomyopathy
DEFINITION
Hypertrophic cardiomyopathy (HCM), the most common genetic cardiac disease, occurs in approximately 1 in 500 young adults.1,2 It is defined as left ventricular (LV) hypertrophy in the absence of an underlying cause, such as systemic hypertension or valvular aortic stenosis.3 In the obstructive form of HCM, septal hypertrophy and abnormal systolic anterior motion of the mitral valve combine to produce left ventricular outflow tract (LVOT) obstruction and variable degrees of mitral valve regurgitation; outflow tract obstruction in HCM is distinct in morphologic appearance and prognosis from congenital membranous subaortic stenosis.4 In addition, some patients will have symptoms due to mid-ventricular obstruction. The condition is important for surgeons because obstruction may occur in more than 70% of patients with HCM,5 and transaortic septal myectomy is highly effective in the management of hypertrophic obstructive cardiomyopathy (HOCM).
HISTORICAL NOTE
The modern description of HCM is credited to Robert Donald Teare, a London pathologist, who in 1958 likened the pathologic findings to “a tumor of the heart” and recognized asymmetrical hypertrophy and myocyte disarray as a familial condition associated with premature and sudden death in young people.6 In a subsequent paper in 1960, Teare proposed the term obstructive cardiomyopathy.7 In 1957, Brock8 described a 58-year-old woman with breathlessness, angina, and syncope who was referred with the diagnosis of aortic valve stenosis. At operation, the valve was normal, and subaortic muscular obstruction was found. Despite attempts to relieve this with dilators, the patient had a residual gradient above 90 mm Hg and died postoperatively. An autopsy confirmed muscular subvalvar stenosis 2.5 cm below the aortic valve, but the histology was never defined.
At the same time, Bercu and colleagues9 described the familial occurrence of unexplained ventricular hypertrophy simulating aortic stenosis as “pseudo–aortic stenosis.” Working at the National Institutes of Health, Morrow and Braunwald reported “functional aortic stenosis,” and the genetic nature of the disease was suggested by Brent and colleagues, who reported two families with “familial muscular subaortic stenosis” compatible with transmission by a mendelian dominant gene.10
HOCM was the term applied to the condition by Goodwin,11 Holman, and others; Braunwald and associates12 popularized the term idiopathic hypertrophic subaortic stenosis. Asymmetrical septal hypertrophy previously was considered to be specific for the disease,13,14 but it is now recognized that asymmetrical septal hypertrophy is simply a variant of HCM in which there is a predominance of septal hypertrophy. In current practice, the broad definition, HCM, is generally accepted, recognizing that not all patients have the same distribution of hypertrophy and that LVOT obstruction is not present in all phases of the disease or in all patients.
Surgeons were aware of HCM after Brock’s reports and the publication by Kirklin from the Mayo Clinic in 1958.8,15,16 Effective treatment of outflow tract obstruction began with a simple myotomy, an incision in the hypertrophied septum as described by Cleveland and others.17–19 Subsequently, septal myectomy was used to relieve outflow tract obstruction, and this approach has been favored at our clinic since the earliest reports by John Kirklin.16 Barratt-Boyes and associates advocated excision of the hypertrophied septum through a combined approach with incision in the left ventricle and the aorta.20 Other approaches to septal myectomy include the left atrial approach with exposure of the hypertrophied septum after division of the anterior leaflet of the mitral valve21; Lillehei used a similar method but detached the base of the mitral leaflet to expose the septum.22 Swan described the use of a corkscrew to excise septal muscle from a limited LV approach,23 and Julian used a curvilinear LV incision that detached the lower part of the free wall of the septum to expose the septal bulge.24
Thickness of the hypertrophied septum can be reduced by shaving the right ventricular side through right ventriculotomy,25,26 but this technique is less direct than the transaortic approach. Because systolic anterior motion of the mitral valve is an integral part of the mechanism of LVOT obstruction, excision of the mitral valve and replacement with a low-profile prosthesis as described by Cooley is an effective method of relieving outflow tract obstruction.27 This, however, leaves the patient with potential hazards of a prosthetic valve. Rarely, an apicoaortic conduit has been used to relieve complex forms of LVOT obstruction due to HCM.28
All of these earlier methods have been largely replaced by the more predictable and complete transaortic myectomy advocated by Morrow,29 and this is the basis of the extended septal myectomy, the surgical approach we favor that is described here.
CLASSIFICATION
Morphology
Distribution of Hypertrophy
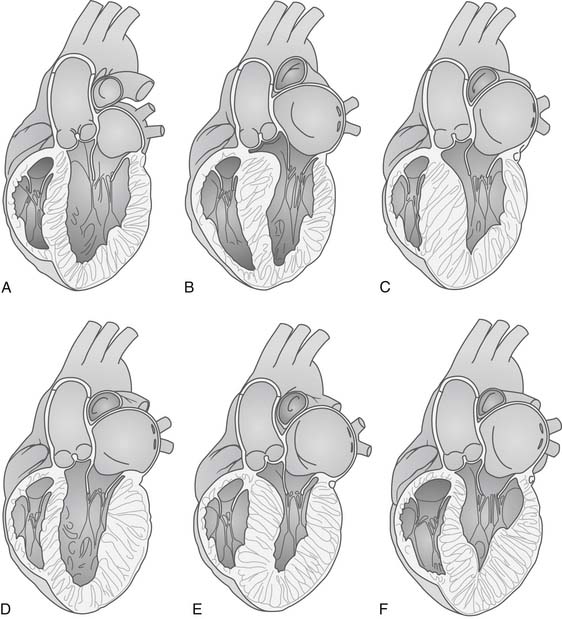
Figure 96-1 shows the variable distribution of LV hypertrophy in patients with HCM. Classically, the anterior basal septum is thickest just cephalad to the anterior leaflet of the mitral valve in its open position. This subaortic obstruction may be accompanied by diffuse ventricular hypertrophy (Fig. 96-1E), or the hypertrophy may be limited to the septum (asymmetrical septal hypertrophy; Fig. 96-1B). In other patients, there may be obstruction in the mid ventricle where hypertrophy of the papillary muscles leads to obstruction of ejection of blood; in other patients, there may be an apical distribution of hypertrophy (Fig. 96-1F). The phenotype of the disease and the appearance and severity of hypertrophy do not correlate with genotype; multiple ventricular morphologic appearances may be found in the same family.
Mitral Valve
In the classic form of HOCM, the mitral valve, especially the anterior leaflet of the valve, moves anteriorly during systole. The posterior leaflet closes against the mid- and free-edge third of the anterior leaflet instead of at the free edge as occurs normally. The free edge of the anterior leaflet is then displaced upward and narrows the LVOT. In addition, the anterior displacement of the valve leaflets produces mitral regurgitation of variable degree, which is typically directed posterolaterally.30 Mitral regurgitation is an important pathophysiologic component of HOCM and contributes to the symptoms of dyspnea and fatigability. Apposition of the anterior leaflet to the bulging septum produces a contact lesion, the whitish endocardial scar, which is useful in guiding septal myectomy. The occurrence of systolic anterior motion is often dynamic, and provocative maneuvers such as Valsalva, squatting, exercise, and, in some cases, adrenergic stimulation may be necessary to elicit systolic anterior motion, mitral regurgitation, and LVOT obstruction. The mechanism of systolic anterior motion is debatable, and proposed mechanisms include the Venturi (pull) effect31,32 and a drag (push) effect.33,34
Anomalies of papillary muscles are present in 15% to 20% of patients with HCM who undergo myectomy.35 These abnormalities include anomalous papillary muscles, direct insertion of papillary muscles into the anterior mitral valve leaflet (Fig. 96-2), fusion of the papillary muscle to the ventricular septum or LV free wall, and accessory muscles and accessory anomalous chordae (false cords). In most cases, these abnormalities do not complicate myectomy or contribute to outflow tract obstruction; but in some patients, anomalous papillary muscles, especially those that insert directly into the anterior leaflet, can contribute to outflow tract obstruction.36

Right Ventricle
Right ventricular wall thickening has been documented in approximately one third of patients with HCM, and 10% have extreme right ventricular wall hypertrophy (≥10 mm).37 Right ventricular hypertrophy, however, rarely leads to fibrosis as evidenced by hyperenhancement on magnetic resonance imaging, and right ventricular outflow tract obstruction due to infundibular narrowing is uncommon38,39 and mainly limited to cases of HCM manifested in children and young adults.40 Right ventricular hypertrophy also occurs as a result of pulmonary hypertension from elevated LV end-diastolic pressure.
Aortic Valve
In contrast to congenital membranous subaortic stenosis,41 LVOT obstruction in HCM does not directly affect the aortic valve. Aortic valve regurgitation has been observed after transaortic myectomy as a result of iatrogenic injury.42
Coronary Arteries
Atherosclerotic coronary artery disease is present in approximately 5% to 15% of patients with HCM, depending on the population studied. At the Mayo Clinic, significant coronary artery disease was detected in half of patients who were selected for coronary angiography, and disease was severe in 26%.43 Survival of HCM patients with obstructive coronary artery disease is reduced compared with that of HCM patients without coronary artery disease, and survival is also poorer than that of patients without HCM who have comparable coronary artery disease and normal ventricular function.
Muscle bridging of the left anterior descending coronary artery (LAD) is not uncommon, occurring in 15% of patients with HCM undergoing coronary angiography at our clinic.44 It is unclear whether bridging of the LAD plays a pathophysiologic role in the disease and associated symptoms of angina; but for adult patients, risk of death and, in particular, of sudden cardiac death is not increased among patients with HCM with myocardial bridging. In contrast, Yetman and coworkers45 reported that among children with HCM, systolic compression of LAD was present in 285 cases and was associated with a greater incidence of chest pain (60% versus 19%; P = .04), cardiac arrest with subsequent resuscitation (50% vs 4%; P = .004), and ventricular tachycardia (80% vs 8%; P < .001). Myocardial ischemia was postulated to be the cause of this poor outcome.
Histopathology
Myocardial Fiber Disarray
A characteristic histologic feature of HCM is myocardial fiber disarray, which consists of short runs of severely hypertrophied fibers interspersed by connective tissue. Myocytes show large, bizarre nuclei with degenerating muscle fibers and fibrosis. It is the disorganized whirling of muscle fibers that is characteristic of HCM. Myocardial disarray is present in the ventricular septum and in the LV free wall, but it is not pathognomonic for HCM. Fiber disarray may be present in myocardium in any conditions in which there is pressure overload, but the proportion of myocardial disarray is much greater in HCM.46,47
Myocyte disarray has been associated with systolic dysfunction, ventricular dilation, and congestive heart failure.48 It may also be a substrate for ventricular arrhythmias. Disarray has also been greatest in hearts with only a mild degree of LV hypertrophy or with absence of a subaortic mitral valve contact lesion, but there seems to be no direct relationship between myocyte disarray and fibrosis or small vessel disease.49
Interstitial Fibrosis
Interstitial fibrosis is an important histologic feature that is present in variable degrees in the ventricle of patients with HCM.50,51 It is important not only as a mechanism of diastolic dysfunction but also because fibrosis detected by magnetic resonance imaging has been associated with arrhythmogenesis and risk of sudden cardiac death.52,53
GENETICS
HCM has a prevalence of 0.2% for phenotypically expressed disease.54 Approximately 60% to 80% of cases are familial, and the remainder result from de novo mutations.55 HCM is genetically diverse, involving sarcomeric and nonsarcomeric proteins, and hundreds of mutations in more than 15 genes have been identified; indeed, double and compound heterozygosity and homozygosity have been reported.56
Most patients with familial disease have mutations in three protein-encoding genes, β-myosin heavy chain (MYH7) in 35% to 50%, myosin-binding protein C (MYBPC3) in 15% to 25%, and cardiac troponin T type 2 (TNNT2) in 15% to 20%. In current practice, the clinical value of genetic testing is uncertain. Van Driest and colleagues57 reported that gene-positive status did not correlate with family history of sudden cardiac death, need for myectomy, or anatomic subtype. Furthermore, specific mutations in HCM are rare; in one study, less than 2% of 293 unrelated patients had putative benign mutations, but serious clinical events developed in these patients, including sudden cardiac death, need for surgical myectomy, and cardiac transplantation.58
Other studies do suggest that certain mutations are associated with poor prognosis. For example, certain mutations in TNNT2, β-tropomyosin (TPM1), and MYH7 may confer increased risk of sudden cardiac death, and more recently, Olivotto reported that identification of myofilament-positive patients through genetic testing predicted a fourfold increase in the risk of the combined endpoint of cardiovascular death, nonfatal stroke, or progression to New York Heart Association (NYHA) class III or class IV symptoms.59 Mutations were associated with more severe systolic and diastolic LV dysfunction, and adverse clinical events occurred irrespective of whether the involved myofilament was thin, intermediate, or thick.
PATHOPHYSIOLOGY
Diastolic Dysfunction
Diastolic dysfunction with elevation of the LV end-diastolic pressure is the principal pathophysiologic finding in HCM. The resulting increase in left atrial and pulmonary venous pressures accounts for the common symptoms of effort dyspnea and limited aerobic capacity. With worsening diastolic function, LV filling becomes more dependent on atrial contraction, and occurrence of atrial arrhythmias, especially atrial fibrillation, can cause an acute and profound decrease in cardiac output and worsening of symptoms.60
Left Ventricular Outflow Obstruction
Surgical treatment of HCM consists primarily of relief of LVOT obstruction. As discussed before, obstruction results from dynamic narrowing of the subaortic area, which in turn is caused by protrusion of the hypertrophied septum in apposition with the anterior leaflet of the mitral valve. Previously, there was debate on the importance of outflow tract obstruction as a mechanism for symptoms in patients with HCM because of the lability of the finding and the question of catheter entrapment when gradients were measured by invasive catheterization.61 It is now recognized that outflow obstruction is much more common than previously thought, correlates importantly with development of symptoms, and may negatively influence long-term survival.
Maron documented resting outflow tract gradients of 50 mm Hg or greater in 37% of 320 patients with HCM and, more important, found exercise-induced gradients (mean 80 ± 43 mm Hg) in an additional 106 patients; thus, as many as 70% of patients with HCM who come to clinical evaluation will have significant outflow tract obstruction.62 Another important finding in this study was the relative unreliability of the Valsalva maneuver (sensitivity 40%) compared with exercise Doppler echocardiography in detecting these dynamic gradients. Latent obstruction can also be documented during hemodynamic catheterization by isoproterenol challenge, and the technique may be useful in patients who are unable to exercise or in patients in whom reliable Doppler echocardiographic signals cannot be measured.63
Mitral Valve Regurgitation
Intrinsic mitral valve disease may contribute to valvular regurgitation in patients with HOCM. Rupture of chordae with resultant leaflet prolapse can precipitate congestive heart failure, and hemodynamics may worsen if HCM is not recognized and patients are managed medically with afterload reduction.64
CLINICAL PRESENTATION AND DIAGNOSTIC CRITERIA
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


