CHAPTER 103 Surgery for Pulmonary Embolism
Pulmonary thromboembolism is a significant cause of morbidity and mortality in the United States and worldwide. The estimated incidence of acute pulmonary embolism is approximately 63 per 100,000 patients in the United States based on clinical and radiographic data,1 and it is related to approximately 235,000 deaths per year based on autopsy data. Acute pulmonary embolism occurs half as often as acute myocardial infarction and is three times as common as cerebrovascular accident. It is the third most common cause of death (after heart disease and cancer).2 Estimates of the incidence of acute pulmonary embolism, however, are generally thought to be low, because in 70% to 80% of patients in whom the primary cause of death was pulmonary embolism, the diagnosis was unsuspected before the death.3,4
Of patients who survive an acute pulmonary embolic event, approximately 3.8% develop chronic pulmonary hypertension.5 Once pulmonary hypertension develops, the prognosis is poor, and this prognosis is worsened in the absence of intracardiac shunt. Patients with pulmonary hypertension caused by pulmonary emboli fall into a higher risk category than those with Eisenmenger’s syndrome and encounter a higher mortality rate. In fact, once the mean pulmonary pressure in patients with thromboembolic disease exceeds 50 mm Hg, the 3-year mortality approaches 90%.6 Therefore, despite an improved understanding of pathogenesis, diagnosis, and management, pulmonary emboli and their long-term sequelae remain frequent and often fatal disorders.
Pulmonary embolism was first described by Laennec in 1819.7 It was he who related the condition to deep venous thrombosis, and Virchow8 later associated the three factors predisposing to venous thrombosis as stasis, hypercoagulability, and vessel wall injury. Virchow distinguished two types of thrombus in the pulmonary arteries of such patients: first, the embolus that arose as thrombus in a systemic vein and, second, the thrombus that occurred in situ in the pulmonary arteries distal to the occluding embolus as a result of the stagnant blood flow in that segment. To prove that pulmonary emboli arose from the peripheral venous circulation, Virchow inserted pieces of rubber or venous thrombi recovered from humans at autopsy into the jugular or femoral veins of dogs. When the animals were sacrificed, the foreign embolic material was found in the pulmonary arteries. Although pulmonary embolism can be caused by tumors, septic emboli, and foreign bodies, the overwhelming majority of pulmonary embolisms result from venous thromboembolism.9
ACUTE PULMONARY THROMBOEMBOLIC DISEASE
Clinical Points
The majority of pulmonary thromboembolic episodes are silent, and it is not until the amount of embolic material is substantial that the patient becomes symptomatic. After an acute major thromboembolic episode, approximately 15% to 20% of patients die within 48 hours.10 In most of the remaining patients, the emboli resolve substantially, by a variety of mechanisms. Therefore, it is in the subgroup of patients who have a sudden fatal outcome (approximately 100,000 annually) that invasive therapy for acute pulmonary embolism might be considered.
In addition to the mechanical factor of pulmonary artery obstruction, there are reflex and hormonal factors that can increase pulmonary vascular resistance at the time of acute pulmonary embolism. Humoral factors, specifically serotonin, ADP, platelet-derived growth factor, and thromboxane, are released from platelets attached to the thrombi,11 and platelet-activating factor and leukotrienes are secreted by neutrophils.12 Anoxia and tissue ischemia downstream from emboli inhibit production of endothelium-derived relaxing factor and enhance release of superoxide anions by activated neutrophils.13 The combination of these humoral effects contributes to enhanced pulmonary vasoconstriction. Thus some patients with a relatively small embolus may have an exaggerated response to the degree of pulmonary vascular obstruction.
In patients without preexisting cardiac or pulmonary disease, an obstruction of less than 20% of the pulmonary vascular bed results in minimal hemodynamic consequences. It is only when the acute pulmonary obstruction exceeds 50% to 60% of the pulmonary vascular bed that cardiac and pulmonary compensatory mechanisms are overcome and cardiac output begins to fall.14 Right ventricular failure occurs, and it is accompanied by systemic hypotension as the amount of blood reaching the left ventricle decreases. The dilated right ventricle causes a shift of the ventricular septum to the left, further compromising left ventricular filling. Although patients with chronic pulmonary artery obstruction can have high pulmonary artery pressure levels that reflect the degree of obstruction, in acute pulmonary embolism the previously normal right ventricle cannot generate these pressures. Therefore, in the case of an acute massive pulmonary embolism, pulmonary artery pressures may be normal, and a pulmonary artery systolic pressure of 30 to 40 mm Hg may represent severe pulmonary hypertension.
Acute pulmonary embolism usually appears suddenly. Symptoms and signs vary with the extent of blockage, the magnitude of humoral response, and the pre-embolus reserve of the cardiac and pulmonary systems of the patient.15 The clinical diagnosis is often missed or falsely made. Most pulmonary emboli occur without sufficient clinical findings to suggest the diagnosis, and in an autopsy series of proven emboli, diagnosis was made in only 16% to 38% of patients while they were still alive.16 The acute disease is conveniently stratified into minor, major (submassive), or massive embolism on the basis of hemodynamic stability, arterial blood gases, and lung scan or angiographic assessment of the percentage of blocked pulmonary arteries.17
For patients with minor pulmonary embolism, physical examination may reveal tachycardia, rales, low-grade fever, and sometimes a pleural rub. Heart sounds and systemic blood pressure are often normal; sometimes the pulmonary second sound is increased. Less than one third of patients with acute pulmonary embolism have concurrent evidence of clinical deep venous thrombosis.16 Room air arterial blood gases indicate a Pao2 of between 65 and 80 mm Hg and a normal PaCO2 of around 35 mm Hg.18 Pulmonary angiograms typically show less than 30% occlusion of the pulmonary arterial vasculature.
Major pulmonary embolism is associated with dyspnea, tachypnea, dull chest pain, and some degree of cardiovascular changes manifested by tachycardia, mild to moderate hypotension, and elevation of central venous pressure.18 Some patients have syncope rather than dyspnea or chest pain. In contrast to massive pulmonary embolism, patients with major embolism (at least two lobar pulmonary arteries obstructed) are usually hemodynamically stable and have adequate cardiac output.19 Room-air-control blood gases reveal moderate hypoxia, with a PaO2 between 50 and 60 mm Hg, and mild hypocarbia, with a PaCO2 of 30 mm Hg or less.18 Echocardiograms may show right ventricular dilation. Pulmonary angiograms indicate that 30% to 50% of the pulmonary vasculature is blocked; however, in patients with preexisting cardiopulmonary disorders, a lesser degree of vascular obstruction may produce similar symptoms.
Massive pulmonary embolism, defined as a pulmonary embolism that causes hemodynamic instability, is truly life-threatening.19 It is usually associated with occlusion of more than 50% of the pulmonary vasculature, but it may occur with much smaller occlusions, particularly in patients with preetxisting cardiac or pulmonary disease. The diagnosis is clinical, not anatomic. Patients develop acute dyspnea, tachypnea, tachycardia, and diaphoresis and may lose consciousness. Both hypotension and low cardiac output (<1.8 L/min/m2) are present. Cardiac arrest may occur. Neck veins are distended, central venous pressure is elevated, and a right ventricular impulse may be present. Room-air blood gases show severe hypoxia (Pao2 <50 mm Hg), hypocarbia (PaCO2 <30 mm Hg), and acidosis.18 Urine output falls, and peripheral pulses and perfusion are poor.
The clinical diagnosis of acute major or massive pulmonary embolism is unreliable and is incorrect in 70% to 80% of patients who have subsequent angiography.20 The differentiation between major or massive pulmonary embolism and acute myocardial infarction, aortic dissection, septic shock, and other catastrophic states can be difficult and costly in time. Although plain chest radiograph, electrocardiogram, and insertion of a bedside Swan-Ganz catheter may add confirmatory information, they will not necessarily prove the diagnosis. Routine laboratory tests are usually normal.
The most common electrocardiographic abnormalities of acute pulmonary embolism are tachycardia and nonspecific ST- and T-wave changes. The major value of the electrocardiogram is in excluding a myocardial infarction. A minority of patients with massive embolism may show evidence of cor pulmonale, right axis deviation, or right bundle branch block.16 Chest radiography may show oligemia (Westermark’s sign) or linear atelectasis (Fleischner lines), both of which are nonspecific findings. Ventilation-perfusion ( ) scans may provide confirmatory evidence, but these studies may be unreliable because pneumonia, atelectasis, previous pulmonary emboli, and other conditions may cause a mismatch in ventilation and perfusion that mimics positive results.21
) scans may provide confirmatory evidence, but these studies may be unreliable because pneumonia, atelectasis, previous pulmonary emboli, and other conditions may cause a mismatch in ventilation and perfusion that mimics positive results.21
In general, negative  scans exclude the diagnosis of clinically significant pulmonary embolism.
scans exclude the diagnosis of clinically significant pulmonary embolism.  scans are usually interpreted as high, intermediate, or low probability of pulmonary embolism to emphasize the lack of specificity but high sensitivity of the test. Pulmonary angiograms provide the most definitive diagnosis for acute pulmonary embolism.22 Using this technique, the diagnosis is established when there are filling defects or obstruction of pulmonary arterial branches, leading to irregular or absent contours or “streaming” of contrast material. Magnetic resonance angiography (MRA) is an excellent noninvasive method for the diagnosis of pulmonary emboli and provides specific information regarding flow within the pulmonary vasculature.23 Unfortunately, this method is expensive, time consuming, and not widely available. Like angiography, it is generally not suitable for hemodynamically unstable patients. Transthoracic echocardiography or transesophageal echocardiography (TEE) with color flow Doppler mapping can provide reliable information about the presence or absence of major thrombi obstructing the main pulmonary artery; however, these techniques are usually inadequate for visualization of the lobar vessels, where the embolic material is often localized. More than 80% of patients with clinically significant pulmonary embolism have abnormalities of right ventricular volume or contractility, often associated with acute tricuspid regurgitation.24 In a subset of patients, abnormal flow patterns can be discerned in major pulmonary arteries during TEE.25
scans are usually interpreted as high, intermediate, or low probability of pulmonary embolism to emphasize the lack of specificity but high sensitivity of the test. Pulmonary angiograms provide the most definitive diagnosis for acute pulmonary embolism.22 Using this technique, the diagnosis is established when there are filling defects or obstruction of pulmonary arterial branches, leading to irregular or absent contours or “streaming” of contrast material. Magnetic resonance angiography (MRA) is an excellent noninvasive method for the diagnosis of pulmonary emboli and provides specific information regarding flow within the pulmonary vasculature.23 Unfortunately, this method is expensive, time consuming, and not widely available. Like angiography, it is generally not suitable for hemodynamically unstable patients. Transthoracic echocardiography or transesophageal echocardiography (TEE) with color flow Doppler mapping can provide reliable information about the presence or absence of major thrombi obstructing the main pulmonary artery; however, these techniques are usually inadequate for visualization of the lobar vessels, where the embolic material is often localized. More than 80% of patients with clinically significant pulmonary embolism have abnormalities of right ventricular volume or contractility, often associated with acute tricuspid regurgitation.24 In a subset of patients, abnormal flow patterns can be discerned in major pulmonary arteries during TEE.25
Prophylaxis
Although prophylactic measures should be considered and used for all patients undergoing major surgery or who have prolonged immobility, certain other patients also fall into a potentially high-risk group for pulmonary embolism. These include patients with previous embolism, malignancy, cardiac failure, obesity, or advanced age.26 The prevalence of deep venous thrombosis of the thigh or pelvis, its strong association with pulmonary embolism, and the identification of the associated risk factors listed earlier provide the basis and rationale for prophylactic anticoagulation for the prevention of acute pulmonary embolism. Simple measures such as compression stockings should probably be prescribed more often and should be used in most nonambulating patients in the hospital. Intermittent pneumatic compression devices are more cumbersome but are also effective. These compression devices are available for the calf or the whole leg. They can provide a range of compression pressures, inflation and deflation duration, and sequential or nonsequential inflation, although a clear difference between these variations has not been demonstrated. Both compression stockings with a compression pressure of 30 to 40 mm Hg at the ankle and pneumatic compression devices reduce the incidence of deep venous thrombosis after general surgery to approximately 40% of control patients.27 Multiple studies have now shown that low-dose subcutaneous heparin or low-molecular-weight heparin given once a day reduces the incidence of deep venous thrombosis28,29 with a concomitant reduction in the incidence of pulmonary embolism. This reduction in thrombosis has not been associated with an excessive risk of bleeding.28 Recent studies suggest that of patients who have deep venous thrombosis diagnosed in the hospital without pulmonary embolism, the probability of clinically diagnosed pulmonary embolism within the next 12 months is 1.7%.30 If pulmonary embolism occurs, the probability of recurrent pulmonary embolism is 6.0%.30 Six months of warfarin anticoagulation is recommended for patients who have deep venous thrombosis with or without pulmonary embolism as prophylaxis against recurrent disease.31,32
Supportive and Thrombolytic Therapy
The majority of patients who die of pulmonary embolism do so within 2 hours of the initial acute event, before the diagnosis can be firmly established, and before effective therapy can be instituted. Once the diagnosis is made, however, treatment will be either medical (supportive and thrombolytic therapy) or surgical (Fig. 103-1).
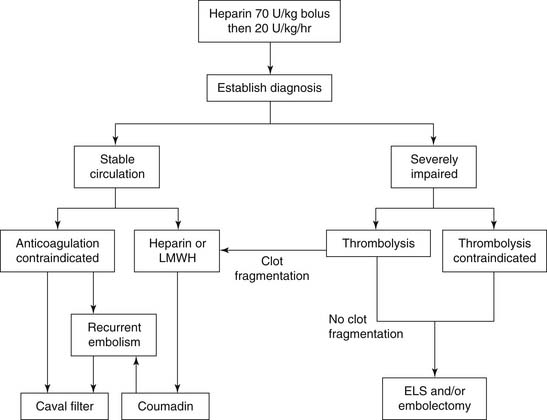
Figure 103–1 Acute pulmonary embolism treatment scheme. ELS, extracorporeal life support; LMWH, low-molecular-weight heparin.
Oxygen should be administered to alleviate hypoxic pulmonary vasoconstriction, and it is likely that a severely affected patient will require intubation and ventilatory support. Pharmacologic agents, including cardiovascular pressors and vasoactive agents, may be instituted to stabilize the patient’s hemodynamics. Once the circulation has been stabilized, both arterial and central venous catheters are placed to monitor cardiac output and pulmonary arterial oxygen saturation. There is debate as to whether pulmonary artery catheters, although clearly helpful in management, should be used in the setting of acute pulmonary embolism, because of the risk of dislodging further thromboembolic material. The electrocardiogram is monitored, a Foley catheter is inserted for accurate recording of urine output, and blood gas levels are obtained. Patients should be heparinized to prevent further propagation of thrombus at its origin and also in the pulmonary arterial tree. If there is no contraindication to anticoagulation, intravenous (IV) heparin is started with an initial bolus dose of 70 U/kg followed by 18 to 20 U/kg/hr. Although heparin prevents propagation and formation of new thromboemboli, it rarely dissolves the existing clot. In most cases, the patient’s intrinsic fibrinolytic system will lyse fresh thrombi over a period of days to weeks.33 Heparin is monitored by measurement of activated partial thromboplastin times, which are maintained between 51 and 70 seconds (roughly twice that of controls), every 6 to 8 hours. The platelet count should be measured every 2 to 3 days to detect the presence of heparin-induced thrombocytopenia. Prothrombin times are also obtained at baseline to prepare for anticoagulation with warfarin later. For the early treatment of stable patients with acute pulmonary embolism, subcutaneous low-molecular-weight heparin (tinzaparin) given once daily has been shown to be as effective and safe as IV heparin with respect to recurrent thromboembolism, major bleeding, and death.34
The natural history of the clot in survivors of acute embolic events is fragmentation and progressive lysis. It therefore follows that the addition of streptokinase, urokinase, or recombinant tissue plasminogen activator (rt-PA, alteplase) improves survival by increasing the rate of lysis of fresh thrombi in the pulmonary arterial tree. Thromboembolytic agents dissolve thrombi by activating plasminogen to plasmin. Plasmin, when in proximity to a thrombus, degrades fibrin to soluble peptides. Circulating plasmin also degrades soluble fibrinogen and, to variable degrees, factors II, V, and VIII. In addition, increased concentrations of fibrin and fibrinogen degradation products contribute to coagulopathy by inhibiting the conversion of fibrinogen to fibrin and by interfering with fibrin polymerization. The currently FDA-approved thromboembolytic agents for the treatment of acute pulmonary embolism are streptokinase, urokinase, rt-PA, and tissue plasminogen activator (t-PA).35 Some new agents (so-called second-generation thrombolytics), such as reteplase, saruplase, staphylokinase, tenecteplase, and antistreplase, are undergoing clinical testing.36
Although thrombolytic therapy has been shown to increase the rate of lysis of fresh pulmonary clots more than therapy with heparin alone,37 there has been little difference measured in the amount of residual thrombus between the two treatments in early studies with small cohorts of patients.38,39 However, two recent trials comparing thrombolytic agents with IV heparin for acute pulmonary embolism have been large enough to detect a significant difference in the most important endpoint—mortality.40,41 More recent experience suggests a trend toward better results with thrombolytic therapy because of a more rapid diminution in right ventricular afterload and dysfunction.37 Compared with heparin therapy alone, thrombolytic agents carry a higher risk of bleeding problems, with up to 20% of patients experiencing a significant bleeding complication.42,43 In general, thrombolytic therapy is contraindicated in patients with fresh surgical wounds, anemia, recent stroke, peptic ulcer, or bleeding dyscrasias.
In select incidents, mechanical removal of pulmonary thrombi has been reported with the use of a catheter device inserted under local anesthesia into the femoral or jugular vein.44–46 Three types of catheters have been introduced into the pulmonary artery with fluoroscopic guidance to remove embolic material. One such device consists of a small terminal cup attached to a flexible catheter.44 Syringe suction is applied to the cup as a thrombus is engaged. The catheter and clot are removed en masse through the venotomy site, and this process may be repeated multiple times. A second reported device uses a rotational pigtail catheter system to mechanically fragment fresh pulmonary emboli under fluoroscopic visualization.45 A variation on this system is the Amplatz catheter that has an impeller housed in a capsule at the tip, driven by a compact air turbine that homogenizes thrombus by maceration and fragmentation. A metal capsule protects the vessel from the rotating impeller.47 A third technique, the rheolytic thrombectomy technique, uses a dual-lumen catheter: the smaller lumen is for delivery of pulsatile pressurized saline, and the larger effluent or exhaust lumen drains the thrombus. High-velocity saline jets are injected toward the effluent lumen. This produces an area of low pressure at the tip of the catheter (the Venturi effect), thereby fragmenting the thrombus and allowing it to be evacuated through the effluent lumen.46 Successful catheter extraction of thrombus with clinically significant reduction in pulmonary arterial pressure varies between 61% and 88%, with the best results achieved for proximal, main pulmonary artery embolism.
Acute Pulmonary Embolectomy
Emergency pulmonary thromboembolectomy is indicated for suitable patients with life-threatening circulatory insufficiency, when the diagnosis of acute pulmonary embolism has been established. If a patient has been taken directly to the operating room without a definitive diagnosis, transesophageal or epicardial echocardiography and color Doppler mapping can confirm or refute the diagnosis in the operating room.48 The primary difficulty with the broad application of operative embolectomy is that it is almost impossible to determine which patients will die without intervention. An emergency pulmonary embolectomy is most feasible (because of time) and most successful in patients who ultimately may not require it, which makes it difficult to establish the efficacy of this operation. Indications for acute surgical intervention include the following: (1) critical hemodynamic condition, with the patient deemed unlikely to survive, (2) definitive diagnosis of pulmonary embolism in the main or lobar pulmonary arteries with compromise of oxygen gas exchange, (3) instability in a patient in whom thrombolytic or anticoagulation therapy is absolutely contraindicated, and (4) the presence of a large clot trapped in the right atrium or ventricle.
Acute pulmonary embolectomy was first described by Trendelenburg49 in 1908, using pulmonary artery and aortic occlusion through a transthoracic approach. There were no surviving patients. Sharp50 performed the first successful open embolectomy, using cardiopulmonary bypass.
As a corollary to this operation, some groups recommend either placement of an inferior vena caval filter or caval clipping before chest closure. Stewart and Greenfield have recommended placement of an inferior vena caval filter under direct visualization before closing the chest.51 Historically, some European surgeons clipped or plicated the intrapericardial vena cava at the end of the embolectomy to prevent migration of lower body clot into the pulmonary circulation; however, this procedure is associated with stasis in the venous system in the lower body, and leg swelling.52 In most centers that offer emergency pulmonary thromboembolectomy, no caval procedure is performed in the perioperative period, and recurrent deep venous thrombosis or pulmonary embolism is treated by anticoagulation with warfarin for 6 months.53 Percutaneously placed Greenfield filters are recommended only for patients with contraindications to anticoagulation or for patients with recurrent pulmonary embolism on therapeutic anticoagulation. The cone-shaped Greenfield filter is most widely used in the United States and is associated with a lifetime recurrent embolism rate of 5% and a lifetime patency rate of 97%.54,55
Extracorporeal Life Support
The wider availability of long-term mechanical perfusion (termed extracorporeal life support [ELS]) using peripheral vessel cannulation to stabilize the circulation offers another approach to life-threatening pulmonary embolism. Most pulmonary emboli, even those that are massive, will dissolve with time. ELS can be instituted outside the operating room setting and can be implemented with rapidity in institutions where preparation for its emergency use has been made. With a trained team, needed equipment, and associated supplies readily available, ELS can be implemented within 15 to 30 minutes.56 For hemodynamic support during the period of a life-threatening pulmonary embolism, venoarterial extracorporeal support can be instituted and maintained for up to several weeks, if necessary.57
This procedure begins with an IV heparin bolus of 1 mg/kg, followed by percutaneous or surgical cutdown of the femoral artery and femoral or internal jugular veins. If pulses are absent or weak, a larger incision is usually faster; however, because patients need heparin and have possibly received fibrinolytic drugs, a minimal wound is preferred. The tip of the venous catheter is advanced into the right atrium to obtain a flow rate of 2.5 to 4.0 L/min using an emergency pump-oxygenator circuit primed with crystalloid.58 The perfusion circuit consists of IV or arterial access tubes, a centrifugal pump, and a membrane oxygenator.59 An electromagnetic flowmeter is placed on the arterial line, and an arterial filter is not applied. While the patient is on ELS, heparin is infused to maintain the activated clotting time between 150 and 180 seconds. In the initial hours after institution of ELS, activated clotting times are measured every 30 minutes, followed by every hour thereafter until decannulation. During the period of ELS, thrombolytic drugs may be instilled directly into the pulmonary artery via a Swan-Ganz catheter to aid in clot lysis. ELS may provide critical support to bridge an unstable patient to surgical embolectomy.60
Results of Acute Pulmonary Embolectomy
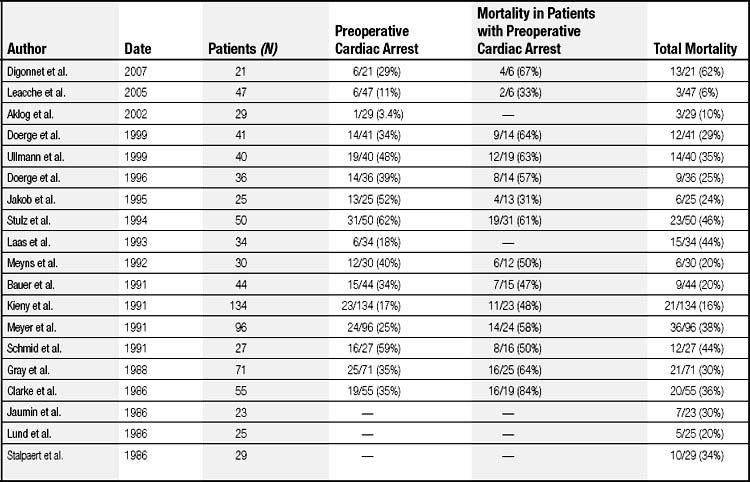
Mortality rates for emergency pulmonary thromboembolectomy vary widely, between 11% and 92% (Table 103-1).53,61–78 It is difficult to compare these studies retrospectively because the operations were performed in different decades, the preoperative hemodynamic states of the patient populations differed, and the treatment plans varied. In general, greater surgical mortality was encountered when a patient had a preoperative cardiac arrest or required ELS support. For example, the mortality in patients who had suffered a cardiac arrest (31% to 84% perioperative mortality) is considerably worse than in those who had not, where mortality is reported between 6% and 44%. Consequently, although some groups report low mortality rates, this may be attributable to the selection of less-ill patients as candidates for surgery. In patients in whom ELS was instituted during preoperative cardiac resuscitation, subsequent operative mortality ranged between 44% and 57%.31,58 Thus the outcome depends largely on the preoperative condition of the patient. Primary causes of death include brain damage, cardiac failure, uncontrollable bleeding, and sepsis. Recurrent embolism after surgery is uncommon,78 and approximately 80% of those who survive surgery maintain normal pulmonary artery pressures and exercise tolerance. In these patients, postoperative angiograms are normal or show obstruction in less than 10% of vessels. A minority of patients who have obstruction in more than 40% of pulmonary vessels after surgery have experienced reduced pulmonary function and exercise tolerance.79
CHRONIC PULMONARY THROMBOEMBOLIC DISEASE
Incidence and Natural History
The natural history of pulmonary embolism is generally total embolic resolution, or resolution leaving minimal residua, with restoration of a normal hemodynamic status. However, for unknown reasons, embolic resolution is incomplete in a small subset of patients. If acute emboli are not lysed in 1 to 2 weeks, the embolic material becomes attached to the pulmonary arterial wall at the main pulmonary artery, lobar, segmental, or subsegmental level.80 With time, the initial embolic material progressively becomes converted into connective and elastic tissue. Often, visualization of the pulmonary arteries by angioscopy a few weeks after unresolved pulmonary embolism reveals vessel narrowing at the site of embolic incorporation. In some patients, some of the pulmonary arterial branches recanalize, with the formation of fibrous tissue in the form of bands and webs.81 By a mechanism that is poorly understood, this chronic obstructive disease may lead to a small-vessel arteriolar vasculopathy characterized by excessive smooth muscle cell proliferation around small arterioles in the pulmonary circulation.82 This small-vessel vasculopathy is seen in the remaining open vessels, which are subjected to long exposure at high flow. Pulmonary hypertension results when the capacitance of the remaining open bed cannot absorb the cardiac output—either because of the degree of primary obstruction by embolus or because of the combination of a fixed obstructive lesion and secondary small vessel vasculopathy.
The incidence of pulmonary hypertension caused by chronic pulmonary embolism is even more difficult to determine than that of acute pulmonary embolism. There are more than 500,000 survivors of symptomatic episodes of acute pulmonary embolism per year.83 The incidence of chronic thrombotic occlusion or stenosis in the population depends on which percentage of patients fails to resolve acute embolic material. One estimate is that chronic thromboembolic disease develops in only 3.8% of patients with a clinically recognized acute pulmonary embolism.5 If these figures are correct and only patients with symptomatic acute pulmonary emboli are counted, approximately 19,000 individuals would progress to chronic thromboembolic pulmonary hypertension in the United States each year. However, because many (if not most) patients with diagnosed chronic thromboembolic disease have no antecedent history of acute embolism, the true incidence of this disorder is probably much higher.
Regardless of the exact incidence, it is clear that acute embolism and its chronic relation, fixed chronic thromboembolic occlusive disease, are both much more common than is generally appreciated and are seriously underdiagnosed. In 1963, Houk and colleagues84 reviewed the literature of 240 cases of chronic thromboembolic obstruction of major pulmonary arteries and found that only six cases had been diagnosed correctly before death. Calculations extrapolated from mortality rates and the random incidence of major thrombotic occlusion found at autopsy supported the postulate that more than 100,000 people in the United States currently have pulmonary hypertension that could be relieved by operation. A recent autopsy analysis of 13,216 patients showed pulmonary thromboembolism in 5.5% of autopsies, and up to 31.3% in older adults.85
Chronic thromboembolic pulmonary hypertension is a disease characterized by an initial thromboembolism to the pulmonary arterial tree that does not resolve itself. Whether this results from an endothelial insult, a preexisting prothrombotic state, or an endothelial progenitor cell trapping and remodeling is the subject of debate. No clear etiology has been defined for the majority of patients who develop chronic thromboembolic pulmonary hypertension. Lupus anticoagulant may be detected in approximately 10% of patients with chronic thromboembolic disease,86 and 20% carry anticardiolipin antibodies, lupus anticoagulant, or both. A recent study has demonstrated that the plasma level of factor VIII, a protein that is associated with both primary and recurrent venous thromboembolism, is elevated in 39% of patients with chronic thromboembolic pulmonary hypertension.87 Analyses of plasma proteins in patients with chronic thromboembolic disease have shown that fibrin from these patients is resistant to thrombolysis in vitro.88 In this study, the fibrin β-chain N-terminus was particularly resistant to thrombolysis, suggesting that it could be responsible for thrombus nonresolution. In addition, there are limited data on the role of thrombomodulin, an endothelial cell membrane surface protein that binds thrombin and acts as a cofactor for the conversion of protein C to activated protein C, a natural anticoagulant in this disease. One group has shown a negative correlation between thrombomodulin plasma concentrations with mean pulmonary arterial pressure and PVR in chronic thromboembolic patients compared with acute venous thromboembolism and control patients.89 In patients who underwent pulmonary thromboendarterectomy, thrombomodulin concentration increased significantly, implying endothelial cell dysfunction in addition to a prothrombotic state in these patients before thromboendarterectomy.
Case reports and small series have suggested links between chronic thromboembolism and previous splenectomy, permanent intravenous catheters, and ventriculoatrial shunts for the treatment of hydrocephalus or chronic inflammatory conditions. In addition to these observations, associations with sickle cell disease, hereditary stomatocytosis, and the Klippel-Trenaunay syndrome have been described.90 However, the vast majority of cases of chronic thromboembolic pulmonary hypertension are not linked with a specific coagulation defect or an underlying medical condition such as was described earlier.
Without surgical intervention, the survival of patients with chronic thromboembolic pulmonary hypertension is poor and is inversely related to the degree of pulmonary hypertension at the time of diagnosis. Riedel and colleagues6 found a 5-year survival rate of 30% among patients with a mean pulmonary artery pressure greater than 40 mm Hg at the time of diagnosis, and 10% in those whose pressure exceeded 50 mm Hg. In another study, a mean pulmonary artery pressure as low as 30 mm Hg was identified as a threshold for poor prognosis.91
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


