Substance Abuse and the Heart
Robert A. Kloner
Shereif H. Rezkalla
Overview
Cocaine may be associated with myocardial ischemia and myocardial infarction (MI). A physician who sees a young person with an unexplained MI should consider cocaine use to be a possible cause. Cocaine may also cause hypertension, cardiomyopathy, arrhythmias, and sudden death. Whether coffee is associated with cardiovascular disease remains highly controversial. However, tea, particularly green tea, may be beneficial. Anabolic steroids and amphetamines have been associated with cases of MI. Excessive alcohol consumption causes dilated cardiomyopathy and hypertension, but moderate consumption of alcohol may reduce coronary artery disease. Tobacco use is unquestionably a strong risk factor for coronary artery disease.
Cocaine Cardiotoxicity
Of the various substances of abuse that may affect the heart, cocaine has received the most attention over the last 10 years (1,2,3,4,5,6,7,8,9). This is largely a result of its widespread use, especially in large urban areas of the United States; the relative ease with which the substance can be obtained; and the occurrence of sudden death among athletes who have used cocaine. In this section, we review the history of cocaine use, describe the basic science of the effect of cocaine use on the heart, detail the results of clinical and autopsy reports of cocaine use in humans, and describe the clinical approach to and therapy for patients who have cocaine cardiotoxicity.
History
Cocaine is an alkaloid from the Erythroxylon coca plant, which grows primarily in South America (10). The Indians of South America have chewed coca leaves for thousands of years. During the time of the Inca empire, coca leaves were used as part of religious ceremonies and given as gifts for special services. In the eleventh century, coca leaves lost their religious significance, and chewing of the leaves spread across all social classes; the leaves were often used as payment for labor in the fields. Writings tell of the restorative powers of coca leaves, including their ability to satisfy hunger and provide a sense of strength and power to the weary. Albert Nieman identified cocaine as the active substance in coca leaves in the late 1850s, and as early as 1880, experimental studies in dogs showed that cocaine had a cardiac-accelerating effect. Sigmund Freud advocated the use of cocaine for a host of ailments, and Carl Koller and William Halstead demonstrated that cocaine had local anesthetic properties.
In the 1860s, a wine known as Vin Mariani, which contained cocaine as a stimulant, was introduced to the market, and in 1886, the drink Coca-Cola was introduced, with cocaine as the active ingredient in the original preparation. Cocaine has since been removed from Coca-Cola, but in the late 1800s,
a number of wines and tonics contained cocaine. The first death associated with cocaine use was reported in the 1890s, and numerous reports of addiction and death—including death from use of cocaine as a local anesthetic—appeared in the early 1900s.
a number of wines and tonics contained cocaine. The first death associated with cocaine use was reported in the 1890s, and numerous reports of addiction and death—including death from use of cocaine as a local anesthetic—appeared in the early 1900s.
That cocaine could affect the human heart was suggested in an article by Lewin in 1931, who stated, “Experiments carried out years ago in Europe proved that drinking of an infusion of 12 g of coca leaves, occasioned—besides a greater frequency of pulse, palpitations of the heart, faintness, seeing of sparks and tinnitus aurium—a feeling of augmented power and a greater desire for activity” (11,12).
By the early 1900s, cocaine was regarded as a dangerous and addictive substance. President William Howard Taft, in an address to Congress in 1910, said that “cocaine posed the most dangerous drug problem that the United States ever faced.” The Pure Food and Drug Act of 1906 required that cocaine be listed as an ingredient whenever it was included in a product, and the Harrison Narcotic Act of 1914 permitted sale of cocaine by prescription only and banned the use of cocaine in patented remedies. Public sentiment was in favor of these regulations, and medical problems caused by cocaine use were relatively infrequent until the 1970s, when cocaine gained prominence as a recreational drug. In the mid-1980s, several highly publicized cases of deaths in athletes who used cocaine and various reports (13) that linked the recreational use of cocaine to cardiac events stimulated renewed interest in the medical consequences of cocaine abuse. (The chewing of coca leaves is still common in South America, where, in general, serious medical consequences have not been reported; this may be because a lower dose of the substance is extracted when the plant leaves are chewed, because cocaine is degraded by gastric acid, and because only a small amount of gastrointestinal absorption occurs when cocaine is taken in this fashion.)
Forms of Cocaine
Cocaine hydrochloride is a water-soluble form of cocaine in which the alkaloid extract of the E. coca plant is dissolved in hydrochloric acid to form a white crystalline powder (14,15). Because the white powder can be absorbed by the mucous membranes, intranasal insufflation (snorting) of this white powder is the most common route of administration. The powder also can be injected intravenously. Cocaine is not readily absorbed gastrointestinally, and the low pH of the gastric mucosa may inactivate the alkaloid (14,15).
Crack cocaine is produced by mixing cocaine powder with baking soda and water and then heating the results. The alkaloid base precipitates into a soft mass that hardens and is then smoked. The term “crack” derives from the cracking or popping sound heard when the cocaine crystals are prepared in this fashion.
Freebase cocaine is made by mixing the cocaine powder with a base (such as ammonia) and a solvent (such as ether). The alkaloid base or “free base” is extracted from the ether by evaporation. Like crack, freebase cocaine is smoked. The flammability of ether poses yet another danger in the preparation of this form of cocaine. Freebase use has declined as crack has become more available and because crack does not pose the problem of flammability.
An estimated 90% of cocaine use in the United States is carried out through nasal snorting. In addition, approximately one third of cocaine users have smoked crack. Less than 10% have injected the drug intravenously. Because cocaine is rapidly absorbed through the respiratory tract, smoking of either crack or freebase cocaine rapidly delivers cocaine to the circulation. Cocaine enters the brain within 6 to 8 seconds by the respiratory route (smoking) and within 10 to 15 seconds by an intravenous route. Snorting cocaine results in peak cocaine concentrations in 30 to 60 minutes (15). The amount of cocaine that can be absorbed through the nasal route may be self-limited to some extent because of vasoconstriction of the nasal mucosa induced by cocaine itself.
Cocaine is often combined with other substances, most commonly ethanol. An estimated 9 to 12 million individuals use this combination on a recreational basis (16,17). Cocaethylene is the cocaine metabolite formed in the presence of ethyl alcohol and may be more potent than cocaine itself (see section on cocaethylene).
Pharmacology
The pharmacology of cocaine is complex and has been the subject of a vast amount of basic science research during the last 10 years. First, cocaine is a local anesthetic. It blocks sodium and potassium channels (6) and thus blocks initiation and conduction of electrical impulses. This effect accounts for prolongation of electrocardiographic (ECG) intervals and decreases in cardiac contractility and may lead to a proarrhythmic effect (18). Second, cocaine blocks the reuptake of neurotransmitters at the presynaptic endings of nerves. As a result, neurotransmitters such as dopamine and norepinephrine accumulate in the synaptic cleft, which results in an intense sympathomimetic effect (6). This second major action of cocaine accounts for increases in heart rate, blood pressure, and contractility, and, by stimulation of α-receptors in the vasculature, contributes to vasoconstriction. A third pharmacologic action of cocaine that affects the cardiovascular system is a decrease in vagal tone, which also contributes to an increase in heart rate (19). Thus, on one hand, cocaine may have a cardiodepressive effect resulting from its effect on sodium and potassium channels. On the other hand, it has a stimulatory effect because of its sympathomimetic properties. These opposing forces may result in very complex outcomes (20).
Effects of Cocaine Administration in Experimental Studies
Cocaine directly depresses the myocardium in experimental studies, resulting in both regional and global abnormalities (21,22,23,24,25,26,27,28,29,30,31,32,33,34,35). When administered intravenously to animals, it results in various degrees of conduction abnormalities and lethal ventricular arrhythmias (36,37,38,39,40,41,42,43,44,45,46,47,48,49). It results in significant myocardial ischemia because of its deleterious effects on platelet function (46,47,48) and the coronary arteries (49,50,51,52,53,54). Details of the various laboratory studies are summarized elsewhere (2).
Controlled Studies of Administration of Cocaine in Humans
Cocaine is known to increase both heart rate and blood pressure in awake individuals. Fischman and coworkers (55) demonstrated a dose-related increase in these parameters when patients received intravenous cocaine. Although small doses did not have a significant effect, an intravenous injection of cocaine, 16 and 32 mg, resulted in a significant rise in both heart rate and blood pressure in human volunteers. The changes peaked at 10 minutes after administration and returned to baseline in approximately 1 hour.
To examine the acute effects of cocaine in humans, investigators administered low doses (2 mg/kg) of intranasal cocaine
to patients in the cardiac catheterization laboratory (3,56,57,58). They observed increases in heart rate and blood pressure but mild, diffuse reductions in coronary caliber (8% to 12% of normal), a 33% increase in coronary vascular resistance, and a 17% reduction in coronary sinus blood flow. Thus, even though myocardial oxygen demand increased—with an increase in double product—these patients exhibited a decrease in coronary blood flow. It is likely that in cocaine abusers, who often use much higher doses than those administered in this controlled study, the imbalance in oxygen supply and demand is even worse. It was shown that the cocaine-induced coronary vasoconstriction could be reversed with the α-adrenergic blocking agent phentolamine, suggesting that α-receptor stimulation by cocaine is a crucial aspect of the vasoconstriction. The vasoconstriction was worse at sites of atherosclerosis and could be relieved by nitroglycerin (59). The β-blocker propranolol potentiated the cocaine-induced vasoconstriction, presumably because the β-receptors were blocked, leaving the α-receptors unopposed. Investigators showed (60) that accumulation of cocaine metabolites may also contribute to coronary vasospasm. Addition of ethanol to intranasal cocaine did not worsen coronary vasospasm (16). Isolated human coronary arteries from patients undergoing cardiac transplantation have also been shown to undergo vasospasm when exposed to cocaine.
to patients in the cardiac catheterization laboratory (3,56,57,58). They observed increases in heart rate and blood pressure but mild, diffuse reductions in coronary caliber (8% to 12% of normal), a 33% increase in coronary vascular resistance, and a 17% reduction in coronary sinus blood flow. Thus, even though myocardial oxygen demand increased—with an increase in double product—these patients exhibited a decrease in coronary blood flow. It is likely that in cocaine abusers, who often use much higher doses than those administered in this controlled study, the imbalance in oxygen supply and demand is even worse. It was shown that the cocaine-induced coronary vasoconstriction could be reversed with the α-adrenergic blocking agent phentolamine, suggesting that α-receptor stimulation by cocaine is a crucial aspect of the vasoconstriction. The vasoconstriction was worse at sites of atherosclerosis and could be relieved by nitroglycerin (59). The β-blocker propranolol potentiated the cocaine-induced vasoconstriction, presumably because the β-receptors were blocked, leaving the α-receptors unopposed. Investigators showed (60) that accumulation of cocaine metabolites may also contribute to coronary vasospasm. Addition of ethanol to intranasal cocaine did not worsen coronary vasospasm (16). Isolated human coronary arteries from patients undergoing cardiac transplantation have also been shown to undergo vasospasm when exposed to cocaine.
Administration of cocaine as a local anesthetic agent for use in laryngoscopy increased the frequency of ventricular premature beats (61). There have been occasional case reports of acute MI when intranasal cocaine was used as a topical anesthetic for nasal operative procedures. The effects of cocaine on human fetal left ventricles were studied (62). Cocaine reduced action potential amplitude, depressed the force of ventricular contraction, and, then, at 90 minutes of exposure, resulted in electrical and mechanical arrest, which also suggests that cocaine has a direct cardiodepressant effect on cardiac muscle. Finally, we observed that when human blood is exposed to cocaine, in approximately one half of the samples, platelet aggregation to adenosine diphosphate but not collagen or epinephrine is increased (Fig. 36.1) (47). Further studies showed that volunteers receiving cocaine at a dose of 2 mg/kg intranasally had platelet activation, α-granule release, and formation of platelet microaggregates (48).
In summary, controlled studies in humans receiving low doses of intranasal cocaine showed that cocaine increases heart rate and blood pressure but causes coronary vasoconstriction at the same time. Thus, the potential for ischemia through an imbalance between oxygen supply and demand may occur, especially when even higher doses of cocaine are used.
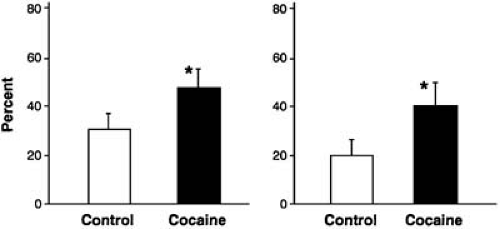 FIGURE 36.1. Platelet aggregation from humans (1 mg, adenosine diphosphate) after incubation with saline or cocaine. Peak aggregation (left) and aggregation at 15 minutes (right). Data are expressed as mean ± standard error of the mean; *p < .02. (From Rezkalla SH, Mazza JJ, Kloner RA, et al. Effects of cocaine on human platelets in healthy subjects. Am J Cardiol 1993;72:243–246. ) |
TABLE 36.1 Cardiovascular complications of cocaine use | |
|---|---|
|
Cardiovascular Disease Associated with Cocaine Use
Most of our knowledge regarding the clinical complications of cocaine use in patients comes from clinical reports and autopsy studies. A number of cardiovascular diseases associated with cocaine use have been reported in the literature (63,64). These are summarized in Table 36.1, and mechanisms of cocaine action are illustrated in Figure 36.2.
Acute Myocardial Infarction
Four possible causes of MI are related to cocaine use. (a) Coronary artery spasm is caused by the intense α-sympathetic stimulation associated with use of cocaine. (b) Thrombus formation was observed in 24% of patients in whom coronary artery disease was absent (65). Thrombus may have occurred on top of an area of vasospasm that was then relieved, and increased platelet aggregability may contribute (47). Thrombus formation may also occur on top of atherosclerotic narrowing. Plaque fracture or rupture, which is common in most Q-wave MIs, is usually not observed in cocaine-related infarcts (53,66). (c) Increased myocardial oxygen demand may occur in the presence of a fixed lesion. Chronic use of cocaine may cause an acceleration of atherosclerosis, resulting in atherosclerotic narrowing in young patients (53). The sympathomimetic responses of increased heart rate and blood pressure, in addition to the stenosis, may contribute to infarction. (d) Some combination of the foregoing three factors may cause MI.
More than 100 cases of cocaine-related MI have been reported in the literature (67). Many more probably occur but are never reported. Clinical, angiographic, and autopsy features reveal some unique properties of cocaine-related infarction. MI after cocaine use typically occurs within 3 hours of use, but this period ranges from minutes to a few days. The median time of onset of chest pain in the infarct patient depends on the route of administration (65). Onset of chest pain occurred at a median time of 30 minutes with intravenous use, 90 minutes with use of crack cocaine, and 135 minutes with nasal insufflation. In a study of 3,946 patients with acute MI (68), 38 patients reported cocaine use before the clinical presentation. The risk appears to be higher in the 60 minutes after cocaine administration, and it rapidly decreases after the first hour. In one study, the description of chest pain was typical: “pressure, crushing, squeezing, tightness, discomfort, heaviness, and in one case, sharp.” Forty-four percent of patients had a history of chest pain (65). There may be a vasospastic component to some of these cases, as suggested by Holter monitoring studies of cocaine use (69). In 1988, at Montefiore Medical Center in Bronx, New York, 35 patients were admitted for chest pain associated with cocaine use. Of these, 11 developed infarction (70). Chest pain after the use of cocaine is not always caused by
MI and may indicate the presence of angina or be noncardiac in nature.
MI and may indicate the presence of angina or be noncardiac in nature.
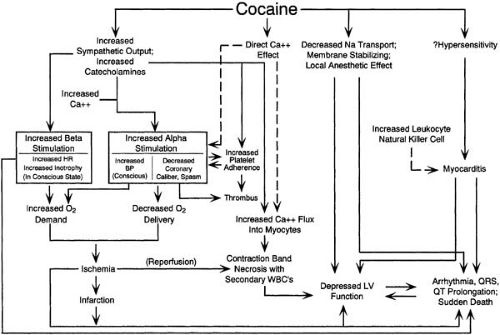 FIGURE 36.2. Schematic diagram showing potential mechanisms of the cardiotoxic effects of cocaine. Two primary mechanisms are suggested. First, cocaine inhibits presynaptic reuptake of catecholamines, resulting in a potentiation of the sympathetic nervous system and an increase in catecholamine levels. Second, cocaine inhibits sodium transport across the sarcolemmal membrane, leading to a membrane-stabilizing or local anesthetic effect that has been likened to a type I antiarrhythmic effect. In conscious preparations, increased sympathetic output and catecholamine levels result in an increase in heart rate and, in some studies, inotropy, which can increase oxygen demand. α-Stimulation of blood vessels causes a vasoconstrictor response with increase in blood pressure in conscious preparations, also resulting in increased oxygen demand. α-Sympathetic stimulation decreases coronary artery caliber, increases coronary vascular resistance, and may lead to coronary spasm in both conscious and anesthetized preparations, thus reducing oxygen supply. Repetitive bouts of coronary spasm might alter or damage the endothelium, contributing to accelerated atherosclerosis, which has been reported with use of cocaine. Increased platelet aggregability has been reported with use of cocaine (and may be related to increased catecholamine levels), which can contribute to thrombus formation. All of these factors contribute to an imbalance between oxygen supply and demand and may thus lead to myocardial ischemia with subsequent infarction and associated left ventricular dysfunction and arrhythmias. Increased sympathetic output may also contribute to tachyarrhythmias. Catecholamine excess is known to lead to contraction-band formation, and this is thought to be related to calcium overload. Isner and Chokshi ( Isner JM, Chokshi SK. Cardiac complications of cocaine abuse. Annu Rev Med 1991;42:33–38 ) postulated that cocaine may have a direct effect on calcium flux into blood vessels (and perhaps myocytes), leading to vasoconstriction that may be independent of the sympathetic nervous system (dashed lines). The local anesthetic effect of cocaine may cause direct depression of inotropy (which in some studies surpasses any indirect positive inotropic response caused by sympathetic simulation) and can lead to a transient cardiomyopathic presentation. This local anesthetic effect can also lead to prolongation of electrocardiographic intervals, including QRS and QT duration, perhaps resulting in arrhythmias and sudden death, much like the proarrhythmic effects of agents such as quinidine. Hypersensitivity to cocaine has been postulated but not absolutely proved to be a cause of myocarditis. In addition, an increase in natural killer cell activity (which may be related to increased catecholamines) has been reported with cocaine and could lead to myocarditis. Scattered foci of myocarditis could lead to a cardiomyopathic presentation and form the nidus for arrhythmias. BP, blood pressure; HR, heart rate; LV, left ventricular; WBC’s, white blood cells. (FromKloner RA, Hale S, Alker K, et al. The effects of acute and chronic cocaine use on the heart. Circulation 1992;85:407–419 , with permission of the American Heart Association.) |
In one report, the mean age of patients with acute MI associated with cocaine was 33 years, and 92% of patients were male (70). The most common associated risk factor was cigarette smoking. There does not appear to be any clear-cut dose relationship between the use of cocaine and development of MI. Chronic, occasional, and first-time users may develop infarction. The ECG was abnormal in 90% or more of patients in the emergency department. ECG abnormalities included ST-segment elevation, T-wave inversion, and Q waves. Both Q- and non–Q-wave infarcts have been reported after cocaine use. High creatine kinase (CK) levels may be present as a result of trauma or rhabdomyolysis. Therefore, CK-MB elevation is needed to document damage to the myocardium (71). Cocaine users who develop coronary artery disease and MI tend to be older and to have multiple risk factors, such as smoking, compared with users who do not have evidence of coronary disease (72).
Coronary vasospasm has been well documented in only a few cocaine-related infarct patients. Coronary stenosis is present in 29% of cases. Thrombosis is present in approximately one fourth of cases. In an analysis of 92 cases in which either coronary angiographic or coronary anatomic data
were collected at autopsy (67), 38% of patients had normal coronary arteries. Of those, 77% had infarcts of the anterior wall.
were collected at autopsy (67), 38% of patients had normal coronary arteries. Of those, 77% had infarcts of the anterior wall.
In a review of the literature (65), 22 of 91 cases of cocaine-induced infarction had some complication: 6 had congestive heart failure, 3 had cardiogenic shock, 11 had potentially life-threatening arrhythmias, 3 had ventricular fibrillation, 6 had ventricular tachycardia, and 2 had cardiac arrests. Three deaths occurred. In the series from Montefiore Hospital (70), among 22 patients with cocaine-related acute MI, no in-hospital deaths occurred, and only one episode of heart failure and ventricular tachycardia was seen. A case report of a patient who developed an intraventricular thrombus after cocaine-induced MI was described (73).
Management of Cocaine-Induced Myocardial Ischemia and Myocardial Infarction
No controlled, randomized, prospective trials clarify the best way to manage cocaine-induced ischemia or infarction. As stated earlier, not all episodes of chest pain associated with cocaine are necessarily MIs. The incidence of MIs associated with cocaine-induced chest pain was 0% to 31% in retrospective analysis and approximately 6% in prospective analyses (74). In addition, not all cocaine-related chest pain is necessarily ischemic; pleuritic pain has been reported. Chest radiographs should be obtained to rule out pneumothorax and pneumomediastinum, which have both been reported with cocaine use. Initiation of thrombolytic therapy should not be delayed while chest radiographs are awaited, however, unless physical examination strongly suggests conditions other than MI.
Patients who enter the emergency department with pain that suggests ischemia (but do not have evidence of infarction) after cocaine use should be observed (74,75,76). One report (74) favored a 12-hour observation period during which ECG and serial CK-MB measurements are obtained to rule out MI. Recent studies have further validated this concept (77,78). Total creatine phosphokinase level elevation appears to occur in a significant number of cocaine users. This is an unreliable screening test for the detection of myocardial injury (79). Myoglobin level is not reliable in detecting MI secondary to cocaine use, but troponin-1 and CK-MB appear to have a good specificity (80). If possible, previous ECGs should be obtained because cocaine users not infrequently also have early repolarization variants on their ECG or left ventricular (LV) hypertrophy. In a prospective study, dobutamine stress echocardiography was both beneficial and safe in the evaluation of chest pain in this group of patients (81).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


