Smoking
Joaquin Barnoya
Stanton A. Glantz
Overview
Tobacco smoke, either as active or passive smoking, is a leading cause of preventable cardiovascular diseases (CVD). In the United States, during 1997–2001, smoking was responsible for an annual average of 260,000 deaths among men and 178,000 deaths among women. One third (34.7%) of these deaths where from CVD (1). Ischemic heart disease (IHD) is the leading cause of death from secondhand smoke (SHS), accounting for more than 10 times the number of deaths from lung cancer (1). The effects of tobacco smoke on the cardiovascular system are multiple and reinforce each other. These effects include platelet activation, endothelial dysfunction, inflammation, altered lipid levels and metabolism, and hemodynamic effects. These effects occur rapidly, often within minutes of active or passive smoking.
Cessation of exposure to tobacco smoke leads to a fast decline in the risk associated with exposure. When someone stops smoking, exercise tolerance improves the next day. Half the excess risk of acute myocardial infarction (AMI) is gone in 1 year, most is gone in 3 years (2), and many of the acute effects on the cardiovascular system begin to disappear within hours after ending exposure.
Tobacco control should be viewed from both an individual and societal standpoint. From an individual standpoint, every smoker should be encouraged to quit or not to smoke around others and every nonsmoker should be encouraged to avoid exposure to SHS. Currently there are multiple pharmacologic treatments to help patients quit, and smoke-free environments have been shown to reduce smoking. Every cardiologist should become active in the societal fight against tobacco and SHS.
Epidemiology of Smoking and Exposure to Secondhand Smoke
Tobacco has been used for thousands of years. A Mayan stone carving more than 1,000 years ago displays the first record of tobacco use in human history. In 1884, James Bonsack developed the cigarette manufacturing machine, resulting in the mass production of cigarettes. In addition to this machine, the development in 1892 of the safety matches fueled an increase in cigarette consumption, along with the development of an aggressive increase in cigarette marketing (3). Since then, tobacco became a major public health problem; by 2020 smoking is expected to kill 10 million people per year, most of them (70%) in low- and middle-income countries (4).
Smoking prevalence in the United States has been declining. In 2003, approximately 21.6% (45.4 million) of U.S. adults were current smokers, half the rate in 1950 (44%) (Fig. 8.1) (5). Eighty-one percent of these smokers do so every day. More men than women smoke (24.1% and 19.2%, respectively) and more young than old people smoke (5). In addition, smoking is more prevalent among those living below the poverty level and in those with low educational level compared to those living above the poverty level and the highly educated (5).
The tobacco epidemic has spread worldwide. For example, in China, in 2001, 60% of men and 7% of women smoked, representing approximately 147 million men and 15 million women age 35 to 74 (6).
Most smokers want to quit smoking or cut down. In 2003, 41.1% (15.1 million) current smokers reported they had stopped smoking for at least 1 day during the past 12 months because they were trying to quit (5). Young adults have the highest spontaneous quit rates (7) and quitting before age 35 results in mortality patterns equal to that of nonsmokers (Fig. 8.2) (8). Regardless of age, every smoker should be encouraged to quit and at least be referred to a smoking cessation quit-line.
SHS exposure continues to be a major threat to most of the world’s population. In the United States, only 43% of blue collar workers enjoy smoke-free environments compared to 75% of white collar workers (9). The 2000 National Youth Tobacco Survey found that 4 in 10 students (grades 6–12) live in homes where others smoke and 7 in 10 are exposed to smoke in public places (10). In China, a cross-sectional survey in 2000 and 2001, found that 27% of nonsmoking men and 26% of nonsmoking women were breathing SHS at the workplace (6). In Europe and Latin America, high concentrations of nicotine have been found in bars and restaurants, and in lower concentrations, in hospitals and high schools
(11,12). Many communities, some states in the United States, and several countries (Ireland, Norway, Sweden, New Zealand, Italy and Uruguay as of March 2006) have implemented smoke-free policies, setting an example to follow and protecting their citizens from the dangers of SHS.
(11,12). Many communities, some states in the United States, and several countries (Ireland, Norway, Sweden, New Zealand, Italy and Uruguay as of March 2006) have implemented smoke-free policies, setting an example to follow and protecting their citizens from the dangers of SHS.
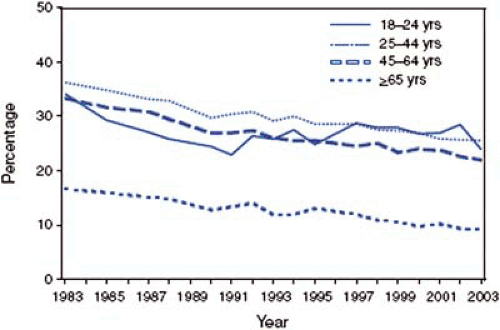 FIGURE 8.1. Smoking prevalence in the United States has declined over the last 20 years. The decline was faster in the first 10 years than in the last 10 years. In addition, most of the decline was among older age groups, the 18- to 24-year-old group has not experienced such an encouraging decline. (Source: Centers for Disease Control and Prevention. Cigarette Smoking Among Adults—United States, 2003. MMWR 2005;54:509–513. ) |
Smoking and Cardiovascular Disease
Smoking is a leading risk factor for all CVD. Coronary heart disease (CHD), stroke, and peripheral artery disease are all increased in smokers compared to nonsmokers. IHD accounts for the largest number of deaths from CVD caused by smoking (64% in men and 60% in women) (1).
CHD, including AMI, IHD, and angina pectoris, risk increases continuously with daily cigarette smoking (13). The relative risk of nonfatal AMI in the 35- to 39-year age group is 4.9 (95% confidence interval [CI] 3.9–6.1) in men and 5.3 (95% CI 3.2–8.7) in women (13). The risk is dose dependent and declines with age (because the baseline risk of heart disease is greater at older age) (14). The fact that the risk is highest in young people highlights the particular relevance of smoking cessation among this group.
Compared to nonsmokers, smokers have a higher risk of sudden death (especially women taking oral contraceptives (15,16), recurrent ischemia after coronary artery bypass grafting (CABG), and reocclusion after an AMI (16). Smokers also have a higher incidence of abdominal aortic aneurysm, peripheral vascular disease, and renal artery stenosis (17,18). Erectile dysfunction has also been found to be associated with active and passive smoking (19) and is markedly improved by smoking cessation (20).
Passive Smoking and Cardiovascular Disease
Passive smoking increases the risk of heart disease by about 30% (14,21,22). Despite the much lower dose of tobacco smoke inhaled by active smokers, the risk (using questionaires to assess exposure) is about one third the increase seen in active smokers (21). Cotinine, a stable metabolite of nicotine, has been used to assess exposure to SHS. Cotinine, unlike questionnaires, provides complete and objective measure of someone’s total recent exposure to SHS. Using this better estimate of exposure based on cotinine, yields higher risk estimates for the effects of SHS than had been determined based on questionnaires; Whincup et al. (23) estimated the risk of CHD with exposure to SHS to be 1.57 (95% CI 1.08–2.28), similar to that found in light (1–9 cigarettes/d) active smokers (Fig. 8.3) (23).
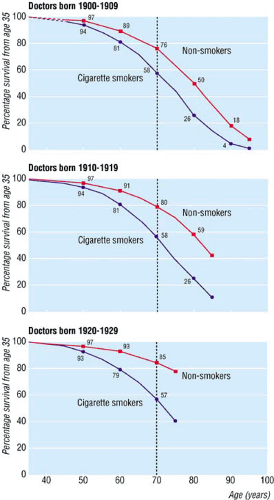 FIGURE 8.2. After smoking cessation there is an increase in survival, regardless of age at quitting. For those who quit before 40 years of age, survival improves almost to that of never smokers and for those who quit before age 35, survival is the same as survival in the never smokers group. (Source: From BMJ. 2004;328(7455):5 with permission from BMJ Publishing group.) |
Effects of Smoking and Passive Smoking on the Cardiovascular System
Active and passive smoking affect the cardiovascular system through the same mechanisms. On average, the biological
effects of passive smoking are nearly as large as the effects of active smoking (21). Table 8.1 summarizes the effects of SHS on the cardiovascular system and compares them to the effects of active smoking. These effects are synergistic and interact with other known cardiovascular risk factors (e.g., diabetes, obesity). In some cases, such as platelet activation and endothelial dysfunction, the effects occur within minutes of exposure to tobacco smoke (24,25).
effects of passive smoking are nearly as large as the effects of active smoking (21). Table 8.1 summarizes the effects of SHS on the cardiovascular system and compares them to the effects of active smoking. These effects are synergistic and interact with other known cardiovascular risk factors (e.g., diabetes, obesity). In some cases, such as platelet activation and endothelial dysfunction, the effects occur within minutes of exposure to tobacco smoke (24,25).
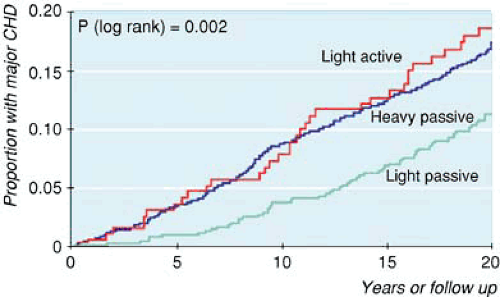 FIGURE 8.3. The risk of heart disease in heavy passive smokers is undistinguishable to the risk in light active smokers using cotinine levels at time zero as the measure of exposure. (Source: Reproduced from BMJ. 2004;329(7439):4 with permission from BMJ Publishing group.) |
Nicotine is the agent in tobacco that has been most widely researched. Even though it is responsible for the addiction, it is not the major agent acting on the cardiovascular system (26). Polycyclic aromatic hydrocarbons (27), oxidizing agents (16), particulate matter (28), and acrolein (29) are more likely the agents that affect the cardiovascular system.
Platelet Activation
Platelet activation leads to thrombosis, a key factor in AMI and sudden death. This prothrombotic state observed smokers partially explains the younger age, lower risk factor prevalence, and less underlying coronary disease observed in smokers compared to nonsmokers at the time of first MI. The immediate increase in the risk of IHD attributable to the increase in platelet aggregation is estimated to be 43% for active smoking and 34% for passive smoking (14).
Tobacco smoke activates platelets through several mechanisms. These mechanisms include endothelial dysfunction, oxidative stress, decrease platelet-derived nitric oxide (NO) production (16), and increased fibrinogen (17) and thromboxane (30). Platelet activation occurs soon after exposure. In active smokers (with established CHD) and passive smokers, platelet activation has been observed 5 minutes after smoking two cigarettes (31) and 20 minutes of breathing SHS (32). Despite the much lower dose of tobacco smoke inhaled by passive smokers, the effect on platelet activation is 96% of that observed in active smokers (21). In addition, side stream smoke (the main component of SHS) has been found to be 1.5 times more potent than mainstream smoke (the smoke inhaled by the smoker) in activating platelets under normal and shear stress conditions (30). “Light” and “mild” cigarette smoke extracts have also been found to be more potent activators than “full flavor” cigarette smoke (33).
The increased platelet aggregability resulting from smoking is ameliorated as early as 2 weeks after smoking cessation (34), suggesting that the effects of tobacco smoke on platelet aggregability are transient and partially reversible.
Endothelial Dysfunction
Endothelial dysfunction is strongly and independently associated with cardiovascular events (35). Endothelial dysfunction results in atherosclerosis, plaque rupture, and decreased blood flow owing to thrombosis and vasospasm. Tobacco smoke exposure (acute or chronic, active or passive) leads to endothelial dysfunction, which is manifest clinically in 15 to 30 minutes (21,25,36). Otsuka et al. (25) exposed healthy smokers and nonsmokers to 30 minutes of SHS at levels comparable to those in a bar. Before exposure, the coronary flow velocity reserve (CFVR, a surrogate marker of endothelial function) was better in nonsmokers than in smokers. After exposure, the CFVR of nonsmokers was undistinguishable from that of smokers. In contrast, smokers experienced no change in CFVR, suggesting that the effect of the smoke constituents was saturated in the smokers (25). Endothelial cell damage has been documented as early as 20 minutes of exposure to cigarette smoke (37,38). A dose–response relationship has also been documented between exposure to tobacco smoke and decreased endothelium-dependent vasodilation (24,39). Evidence suggests that the endothelium partially recovers after exposure has ended (1 year after exposure has ended) (39,40).
NO, secreted by the endothelium and responsible for vessel dilation, is decreased in active and passive smokers (41,42). In animal models with low levels of NO activity, the repair mechanism of the endothelium is impaired (35). Furthermore, the detrimental effects of tobacco smoke on endothelial function have been abolished by adding the NO precursor, L-arginine, to the diet of active and passive smokers (43,44,45).
Atherosclerosis
Lipid levels are altered in smokers and passive smokers (17,21). Tobacco smoke increases low-density lipoprotein (LDL) and decreases in high-density lipoprotein (HDL) (17). In addition to altering lipid levels, cigarette smoke renders LDL more prone to oxidation. Active and passive smokers have higher levels of products of lipid peroxidation and oxidized LDL (17,46). Oxidized LDL is rapidly ingested by macrophages which, in turn, form foam cells in atherosclerotic lesions. Insulin resistance, leading to an atherogenic lipid profile, is also increased by tobacco smoke (16,47).
Active and passive smokers also show evidence of increased inflammatory markers (17,48). Inflammation is now recognized as a key step in the atherosclerotic process (49). Inflammatory markers that have been found to be elevated with tobacco smoke exposure include leukocyte count, acute phase reactants (e.g., fibrinogen, C-reactive protein), interleukin-6, and tumor necrosis factor. The increase observed in passive smokers compared to that observed in active smokers is as or sometimes larger (see Table 8.1). This inflammatory state is reduced after smoking cessation (50).
Increased Oxidative Stress
Smokers and passive smokers have increased markers of oxidative stress (17,21). Under normal conditions, oxidative stress results from free radicals generated during the respiratory process. To protect blood vessels and LDL from oxidation, the body uses antioxidants such as folate, vitamin C, and β-carotene. Smokers and passive smokers have been found to
have lower levels of antioxidants (51,52,53). Therefore, the harmful effects of tobacco smoke are twofold. First, tobacco smoke is a source of free radicals. Second, it leads to a decrease in antioxidant levels that normally would protect the body against oxidative damage.
have lower levels of antioxidants (51,52,53). Therefore, the harmful effects of tobacco smoke are twofold. First, tobacco smoke is a source of free radicals. Second, it leads to a decrease in antioxidant levels that normally would protect the body against oxidative damage.
TABLE 8.1 Comparative Effects of Passive and Active Smokinga | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||



