Sleep-Related Hypoventilation Syndromes
Sleep can have profound changes on the respiratory system (see Chapter 96) especially in patients with underlying cardiopulmonary disorders. Sleep-related hypoventilation syndromes are characterized by an abnormal increase in partial pressure of carbon dioxide (PaCO2) and a decrease in PaO2 during sleep. In an attempt to standardize definitions and facilitate research, in 1999 the American Academy of Sleep Medicine (AASM) included an arterial PaCO2 >45 Torr during wakefulness and a greater than 10 Torr increase in PaCO2 during sleep from awake supine values in the definition of sleep hypoventilation syndromes.1 Similarly, the 2005 International Classification of Sleep Disorders (ICSD-2) incorporated a PaCO2 during sleep greater than 45 mm Hg or disproportionately increased relative to levels during wakefulness in the diagnostic criteria for sleep-related hypoxemia/hypoventilation syndromes.2 More recently, the AASM Sleep Apnea Definitions Task Force revised scoring of sleep hypoventilation in 2012 to include a PaCO2 increase >55 mm Hg for ≥10 minutes or a ≥10 mm Hg increase in PaCO2 during sleep in comparison to awake supine values to a value exceeding 50 mm Hg for ≥10 minutes.3 While based on data that normal individuals rarely have a PaCO2 >55 mm Hg during sleep, the precise PaCO2 level demarcating the transition from physiologic hypercapnia to pathologic hypoventilation remains unclear. The duration of 10 minutes decided by the AASM Task Force was arbitrary and based on consensus with a lack of normative data on the amount of total sleep time at different PaCO2 values in sleeping adults.3 Sleep is associated with stage-specific changes in ventilation covered in Chapter 96. Loss of the wakefulness drive to breathe, altered ventilatory response to hypoxia and hypercapnia and increased upper airway resistance result in a decrease in ventilation in both nonrapid eye movement (NREM) and rapid eye movement (REM) sleep when compared to wakefulness. Consequently there is a small normal physiologic increase in the PaCO2 of 4 to 6 mm Hg during sleep.
The small decrease in ventilation and increase in PaCO2 during sleep is usually of little clinical consequence in normal individuals but in patients with respiratory muscle weakness, altered respiratory mechanics, impaired gas exchange and/or abnormal ventilatory drive, sleep is a vulnerable time. Nocturnal hypoventilation often precedes chronic daytime hypoventilation but the extent to which sleep can elicit and exacerbate chronic hypoventilation is often under appreciated. A high index of clinical suspicion for nocturnal hypoventilation is necessary especially with the availability of effective treatment, and this chapter aims to review some of the common causes of sleep-related hypoventilation.
CAUSES OF SLEEP-RELATED HYPOVENTILATION
The PaCO2 is determined by CO2 production divided by alveolar ventilation (minute ventilation [the product of tidal volume and respiratory rate] minus dead space ventilation). Hypercapnia results when alveolar ventilation is insufficient to meet metabolic needs and is caused by a decrease in minute ventilation, an increase in dead space ventilation, or rarely an increase in CO2 production. The causes of sleep-related hypoventilation can be thought of as disorders of ventilatory drive, that is, “won’t breathe,” or disorders of respiratory mechanics and/or impaired gas exchange, that is, “can’t breathe” (Table 100-1).4 The ICSD-2 classified sleep-related hypoventilation/hypoxemic syndromes by etiology (Table 100-2).
Source: Data from Medicine AAoS. The international classification of sleep disorders: diagnostic and coding manual. 2nd ed. Westchester, IL, 2005.
DISORDERS OF VENTILATORY CONTROL
Disorders of ventilatory drive can result in central apnea and nocturnal hypoventilation. These disorders can be primary including congenital central hypoventilation syndrome (CCHS) and primary idiopathic alveolar hypoventilation where no other cause for central hypoventilation is found. Alternatively these disorders can be acquired, usually following neurologic disorders that affect the brain stem and spinal cord and opiate medications.
CONGENITAL CENTRAL ALVEOLAR HYPOVENTILATION SYNDROME
Congenital central hypoventilation syndrome (CCHS) is a rare cause of central hypoventilation first described in 1970 by Mellins et al.5 CCHS is characterized by hypoventilation and diffuse autonomic nervous system dysregulation (ANSD) but the exact underlying mechanism is uncertain. Studies suggest abnormal brainstem integration of chemoreceptor afferent pathways for ventilation with demonstration of some, albeit blunted, physiologic response of both central and peripheral chemoreceptors. Abnormal central integration of chemoreceptor afferents is also thought to be responsible for the autonomic nervous system dysfunction seen in CCHS. Traditionally considered a disorder of infants and children, with advancements in knowledge and treatment, patients are now surviving into adulthood. Nonetheless, this lifelong disorder is rare. The exact incidence is unknown but a French CCHS registry reported a prevalence in 2005 of 1 in 200,000 live births.6
CCHS is caused by a mutation in the paired-like homeobox 2B (PHOX2B) gene.7,8 The PHOX2B gene on chromosome 4p12 encodes for a transcription factor that plays a role in the development of the autonomic nervous system and regulation of neural crest cell migration. Approximately 90% of PHOX2B mutations involve polyalanine repeat expansion mutations (PARMs) producing genotypes of 20/24 to 20/33 whereas the normal genotype is 20/20. The remaining 10% are heterozygous for nonpolyalanine repeat mutations (NPARMs) and include missense, nonsense, or frameshift mutations. Most expansion mutations occur de novo but in 5% to 10% parents will be mosaic for the PHOX2B mutation, with a 50% chance of transmitting the mutation in all.9
CCHS is a clinically heterogeneous disorder. Studies have demonstrated a relationship between the PHOX2B genotype and CCHS phenotype. A spectrum of hypoventilation severity is seen, and patients with 20/27 to 20/33 genotype and NPARMs typically require 24-hour continuous ventilatory support.10 The syndrome is typically diagnosed in newborns with episodes of cyanosis and apnea usually requiring mechanical ventilation. After maturation of the respiratory and central nervous systems, many patients with CCHS eventually breathe adequately while awake but CCHS does not resolve spontaneously. Hypoventilation in CCHS is most apparent in NREM compared to REM sleep. This is unique from other sleep-related breathing disorders and is thought to be due to intrinsic REM-related ventilatory drive.11 Moreover CCHS children lack perception of dyspnea, do not manifest signs of respiratory distress, and are unable to augment ventilation when faced with a respiratory challenge like an infection or anesthesia; therefore these patients can develop sudden decompensated respiratory failure. Unrecognized, children with CCHS present with complications of chronic hypoventilation including pulmonary hypertension, cor pulmonale, seizures, or developmental delay. While patients can present outside the newborn period with late-onset central hypoventilation syndrome (LO-CCHS), reported as late as 35 years,12 careful review of the medical history typically reveals signs and symptoms compatible with hypoventilation and autonomic dysregulation from the newborn period. It is thought to reflect the variable penetrance of PHOX2B mutations and should be distinguished from rapid-onset obesity with hypothalamic dysfunction, hypoventilation, and autonomic dysregulation (ROHHAD). ROHHAD is not associated with PHOX2B mutations. Patients typically present after the age of 2 years with hyperphagic obesity followed by hypothalamic and autonomic dysregulation, central hypoventilation and behavioral changes, and the syndrome is now considered distinct from CCHS.9,13
ANSD and disorders of neural crest origin are common in CCHS. Hirschsprung disease (a congenital malformation of the enteric nervous system) has been reported in approximately 16% of patients with CCHS and is often of greater severity than the general population with Hirschsprung disease while neural crest tumors including neuroblastoma or ganglioneuroma have been described in 5%.14,15 Other symptoms of autonomic dysfunction described in CCHS patients include diminished pupillary light response, temperature instability, sporadic profuse sweating, altered perception of discomfort and anxiety, and esophageal dysmotility. Cardiac arrhythmias including decreased beat-to-beat heart rate variability, reduced respiratory sinus arrhythmia, and asystoles have been observed.9,16 Children with CCHS may have a characteristic box-shaped facies that is shorter and flatter with a lip trait (Fig. 100-1).17 PHOX2B genotype–CCHS phenotype relationships have also been demonstrated for the more severe CCHS phenotypes that manifest Hirschsprung disease, tumors of neural crest origin, cardiac asystole, ANSD, and facial dysmorphology.9,10 For example, Hirschsprung disease has been reported in 87% to 100% of NPARMs compared to 13% to 30% of PARMs, while neural crest tumors have been described in 50% of NPARMs compared to 1% with PARMs.9
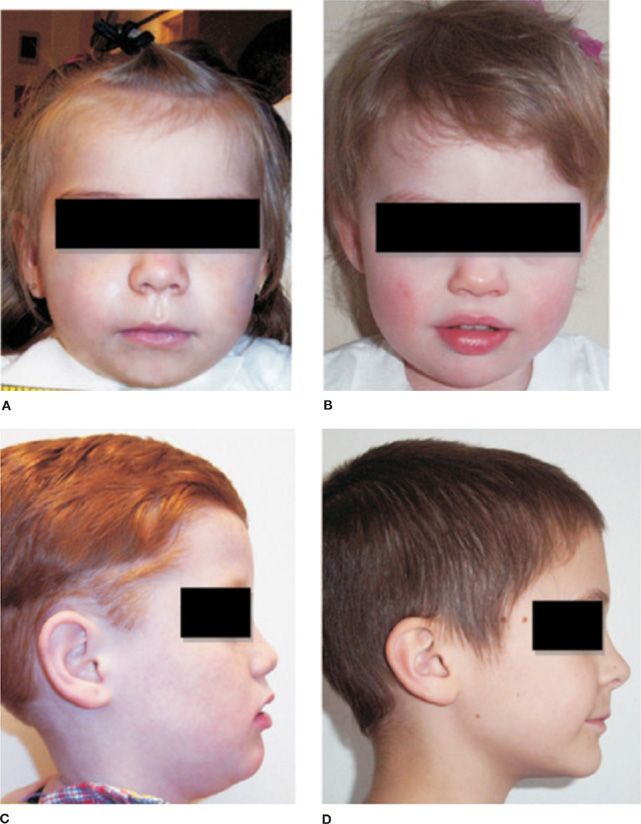
Figure 100-1 Photographs of representative congenital central hypoventilation syndrome (A and C) and control (B and D) subjects showing representative box-shaped facies of CCHS with decreases in the slope of the forehead, upper face height, upper facial inclination, nasolabial angle, upper and lower lip heights, and inferior inflection of the lateral vermillion border of the upper lip. (Reproduced with permission from Todd ES, Weinberg SM, Berry-Kravis EM, et al. Facial phenotype in children and young adults with PHOX2B-determined congenital central hypoventilation syndrome: quantitative pattern of dysmorphology. Pediatr Res. 2006;59(1):39–45.)
Diagnosis of CCHS relies on genetic testing after excluding other causes of central hypoventilation including pulmonary, neurologic, and metabolic etiologies. Genetic testing with PHOX2B Screening Test will identify mutations in 95% of cases. If the PHOX2B Screening Test is negative but there remains a high clinical suspicion the PHOX2B Sequencing Test can be performed to detect NPARM mutations. A sleep study with continuous monitoring of end-tidal CO2 or transcutaneous PCO2 is recommended to assess ventilatory requirements in all stages of sleep. Further diagnostic studies are guided by the clinical evaluation and include but are not limited to screening for other autonomic dysfunction including Hirschsprung with a barium enema or manometry, cardiac arrhythmias with 72-hour Holter monitoring, neural crest tumors with serial chest and abdominal imaging, pupillary function with a comprehensive ophthalmologic examination and neurocognitive testing.
Treatment for CCHS is directed at ensuring adequate ventilation both during sleep and wakefulness ideally with an experienced multidisciplinary team. Chronic ventilatory support is required and in 6% to 33% of patients ventilatory support during both wakefulness and sleep is needed.14 Respiratory stimulants have not been shown to improve ventilatory drive in CCHS. Ventilatory support can be provided by various modalities including positive pressure ventilation via tracheostomy, bilevel positive airway pressure (BPAP) via nasal or full face mask, negative pressure ventilators, and diaphragmatic pacing. The mode of ventilatory support should be guided by the needs of the patient. In infants and younger children positive pressure ventilation via tracheostomy is preferred. When only nocturnal ventilatory support is required, BPAP via a mask interface is commonly used but in patients unable to generate adequate spontaneous breaths to trigger the ventilator, only the timed or spontaneous timed (setting a backup rate) modes guarantee breath delivery. Regardless of the mode of ventilatory support, close monitoring is necessary usually with continuous pulse oximeters and end-tidal CO2 monitors. Apnea/bradycardia monitors will not detect hypoventilation or sinus pauses in CCHS and have no role. With early diagnosis, close monitoring and support, children with CCHS can be expected to function well in society and have a good quality of life but mortality rates of 8% to 38% have been reported.14 Genetic counseling is essential for individuals diagnosed with CCHS.
OBESITY HYPOVENTILATION SYNDROME
The obesity hypoventilation syndrome (OHS) is defined by the presence of obesity (body mass index, BMI ≥30 kg/m2), chronic alveolar hypoventilation with daytime hypercapnia (awake PaCO2 ≥45 mm Hg), and sleep-related breathing disorder in the absence of any other causes of hypoventilation. While first described in ancient Greek literature it was not until the 1950s following case descriptions by Auchincloss and later Burwell that the syndrome gained attention and the term, the “Pickwickian syndrome,” from the character of Joe in The Posthumous Papers of the Pickwick Club by Charles Dickens emerged.18–20
The risk of OHS increases as BMI increases and in the United States it is now estimated that more than one-third of adults are obese.21 As obesity and in particular super obesity (BMI ≥50 kg/m2) increases exponentially, OHS is emerging as a leading cause of sleep-related hypoventilation. A prevalence of 10% to 20% has been reported in obese patients with obstructive sleep apnea (OSA), almost 50% in hospitalized patients with a BMI ≥50 kg/m2, while a conservative prevalence of 0.3% has been estimated in the general US adult population.22–24
 PATHOPHYSIOLOGY
PATHOPHYSIOLOGY
The precise mechanism by which obesity leads to hypoventilation remains incompletely understood. A complex interaction between (1) abnormal respiratory system mechanics and increased work of breathing due to obesity (reduced chest wall and respiratory system compliance, increased airway resistance, expiratory flow limitation, intrinsic positive end-expiratory pressure, reduced respiratory muscle efficiency); (2) sleep-related breathing disorder; and (3) blunted central hypercapnic and hypoxic ventilatory drive, including “resistance” to leptin (a protein produced mainly by adipose tissue that stimulates ventilation) likely exists (Fig. 100-2).23
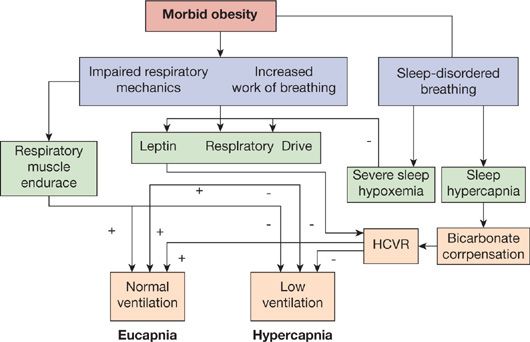
Figure 100-2 Interaction of the various pathophysiologic mechanisms implicated in the development of hypercapnia in the obesity hypoventilation syndrome. (+), positive influence on maintaining ventilation if normal or increased; (–), negative influence on maintaining ventilation if reduced or blunted; HCVR, hypercapnic ventilator response. (Reproduced with permission from Piper AJ, Grunstein RR. Big breathing: the complex interaction of obesity, hypoventilation, weight loss, and respiratory function. J Appl Physiol. 2010;108(1):199–205.)
In approximately 90% of OHS patients the sleep-related breathing disorder is OSA. In the remaining 10% the sleep-related breathing disorder is nonobstructive sleep hypoventilation characterized by sustained hypoxia and an increase in PaCO2 by 10 mm Hg above wakefulness in the absence of significant obstructive apneas or hypopneas. More recently, OHS has been suggested to result from an inadequate compensatory ventilatory response to offload the CO2 that accumulates during apneic and hypopneic events in sleep.26 In addition, if the bicarbonate retained by the kidneys to buffer this transient decrease in pH is slowly eliminated, there is further blunting of the ventilatory responsiveness to CO2, causing progressive hypercapnia (Fig. 100-3).27
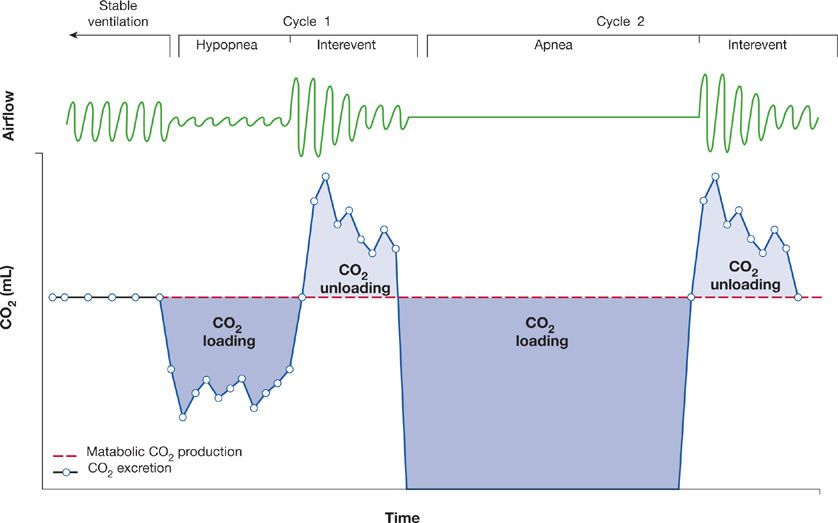
Figure 100-3 Schema depicting a proposed inadequate compensatory ventilatory response to CO2 loading during obstructive respiratory events in the obesity hypoventilation syndrome. The dark-shaded areas depict CO2 loading due to reduced CO2 excretion during respiratory events while the light-shaded areas depict CO2 unloading due to compensatory hyperventilation between respiratory events. (Reproduced with permission from Berger KI, Goldring RM, Rapoport DM. Obesity hypoventilation syndrome. Semin Respir Crit Care Med. 2009;30(3):253–261.)
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
OHS is associated with increased morbidity and mortality, higher rates of hospitalization and ICU admissions, increased healthcare utilization, and reduced quality of life.22–24,28 Timely diagnosis is essential given higher rates of systemic hypertension, angina, congestive heart failure, and cor pulmonale reported in OHS compared to eucapnic obese individuals.23,28 Yet despite the clinical consequences of OHS the diagnosis of OHS is often overlooked and a high index of clinical suspicion is required.24
Typical symptoms of OSA (see Chapter 97) are commonly reported by OHS patients but additional symptoms of hypercapnia including morning headaches may be present. Dyspnea is more frequently reported than eucapnic OSA patients. Pulmonary hypertension, cor pulmonale, and polycythemia are also more common in patients with OHS compared to patients with OSA alone. Not infrequently, OHS patients can present decompensated in acute hypercapnic respiratory failure requiring the ICU.
Physical examination may reveal facial plethora, injected sclera, a prominent pulmonic component of the second heart sound and signs of right heart failure including lower extremity edema in addition to signs suggestive of OSA. Compared to eucapnic OSA patients, OHS patients commonly have awake hypoxemia typically with a PaO2 less than 70 mm Hg. Awake hypoxemia is uncommon in eucapnic OSA patients and a pulse oximetry recording of <94% should prompt consideration of an arterial blood gas measurement.23
 DIAGNOSTIC APPROACH
DIAGNOSTIC APPROACH
The diagnosis of OHS relies on demonstration of awake hypercapnia and an arterial blood gas on room air is necessary. Chronic hypercapnia may be suggested by an elevated serum bicarbonate and a serum bicarbonate of 27 mEq/L or greater has been shown to be highly sensitive (92%) but not specific (50%) for hypercapnia.23,29 Typically patients manifest a >10 Torr increase in their PaCO2 during sleep.
OHS is a diagnosis of exclusion. Other causes of chronic hypoventilation including significant pulmonary disease, chest wall disorders, neuromuscular disease, severe hypothyroidism, and central hypoventilation syndromes must be excluded, but in reality it is likely that the pathophysiologic mechanisms underlying OHS can coexist with these disorders. Further diagnostic testing should be guided by the clinical history and physical examination but should include pulmonary function testing, chest imaging, electrocardiography, thyroid function testing, and a sleep study, where a characteristic pattern is often seen including increased sleep time with SpO2 <90% as outlined in Figure 100-4.23,30,31
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


