Sepsis, Multiple Organ Dysfunction Syndrome, and Chronic Critical Illness
INTRODUCTION
The past four decades have added greatly to our recognition that sepsis, originally defined as a syndrome induced by actively dividing microorganisms in the circulation, is, in fact, a complex and diverse disorder. The term has now come to encompass a constellation of abnormalities that reflect disordered or dysregulated inflammation. As such, sepsis may be initiated by infection; however, it may also occur in the absence of microbial invasion, for example, in response to extensive trauma, uninfected pancreatitis, ruptured aortic aneurysm, or some distinctly unusual entities, such as amniotic fluid embolism. All such patients may develop multiple organ dysfunction syndrome (MODS) and a newly recognized state currently called “chronic critical illness (CCI).” What is now apparent is that our ability to manage shock and to support organ function has unmasked a common family of disorders that have a high mortality and significant morbidity. Familiarity with sepsis is essential for all medical practitioners.
This chapter defines the clinical findings that constitute sepsis, MODS, and CCI. The disorders are described as a function of their position on a continuum of clinical, organ system–specific, cellular, and, indeed, even subcellular, changes that include abnormal biochemistry, metabolism, and energetics. Several pathogenic hypotheses, management strategies, and intriguing new forms of therapy are addressed.
DEFINITIONS, NATURAL HISTORY, AND EPIDEMIOLOGY
The characteristic response to inflammatory stimuli, including surgery and trauma, has been referred to as the stress response, the evolutionary importance of which lies in facilitation of survival and tissue repair.1
Initially in the stress response, an orchestrated neuro-hormonal-humoral mechanism directs substrate delivery to the most vital organs—the heart and brain. Enabling of the mechanism requires rapid development of vasoconstriction, fluid retention, and translocation of intracellular water into the vasculature. In the absence of exogenous life support, death from shock ensues when these endogenous mechanisms are inadequate.
Resuscitation from the initial phase of shock is followed by a period during which cardiovascular function and global metabolism are markedly enhanced.2 The driving force behind this second phase is repair of damaged tissue, with white blood cells serving as the primary effectors of the process.3 To support the increased white blood cell mass, substrate is mobilized from endogenous sources and glucose reserves are rapidly depleted. Because white blood cells are obligate glucose users, muscle (both skeletal and smooth) is broken down to provide precursors for increases in hepatic gluconeogenesis. Nonglucogenic amino acids are used to synthesize structural proteins and enzymes. Energy to support the liver, heart, and other organs is derived from fat and amino acids, since utilization of glucose by tissues other than blood cells and neurons is blocked. Generalized capillary recruitment and leak allow substrate delivery to the avascular area where tissue is damaged. The amount of fluid in the extracellular compartment, particularly in the extracellular, extravascular matrix, increases dramatically. Continued fluid retention and movement of water out of cells fills the dilated, leaky vasculature. Vasodilatation is accompanied by an increase in cardiac output (CO), which further facilitates delivery of substrate. By the fourth day following injury or surgery, neovascularization of damaged tissue results in a sharp increase in substrate delivery to damaged areas. This change is accompanied by a decrease in capillary leak, generalized increase in vascular tone, and mobilization and excretion of fluid in the matrix. Water also returns to cells. In most cases, patients recover uneventfully.
In an unknown fraction of cases, the inflammatory process becomes disordered. Initially the process was defined by the presence of two or more of the criteria listed in the original definitions of sepsis (Table 142-1)4 which indicated enhanced inflammation, a syndrome referred to as the systemic inflammatory response syndrome or SIRS. Use of SIRS criteria has been very helpful in enabling early recognition of patients for inclusion in epidemiologic and interventional studies on sepsis. However, this approach, even when updated,5 has been increasingly recognized as problematic because it is too nonspecific and may reflect a view of septic pathophysiology that is too limited. Recent data have shown that disordered inflammation also includes “immunoparalysis,” with white blood cell function that is also markedly diminished.6,7 Thus, virtually all aspects of inflammation are altered in early sepsis.
Source: Data from Levy MM, Fink MP, Marshall JC, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003;31(4):1250–1256.
Previous descriptions of sepsis detailed subsequent development of a state of organ dysfunction or failure.8–11 This syndrome, MODS, is now considered to begin quite early, perhaps at the very beginning of immune dysregulation. While MODS rarely leads to death, it does progress to a persistent, prolonged, and unchanging state that has come to be known as CCI. Recent studies of CCI indicate that the subset of patients who remain in the ICU for greater than 30 days account for only 8% of total admissions but for 48% of total occupied beds. Six months after hospital discharge, 50% of the cohort is still alive and 80% of the survivors are living in their previous place of residence.12
Data on incidence and outcome in sepsis are difficult to determine because the scientific community has used varying definitions of sepsis. In a study published in 2001, sepsis was noted as responsible for 215,000 deaths each year, and overall hospital mortality rate was 28.6%.13 Expenditures associated with caring for patients with sepsis was estimated at $16.7 billion. More recent work based on examination of the Medicare database from 1996 to 2008 indicated that the number of patients with a diagnosis of sepsis had increased to nearly a million annually.14 Indeed, the incidence of sepsis in elderly patients has become epidemic: 60% of patients who develop sepsis and 75% of the deaths in sepsis are in patients older than age 65 years. These data are consistent with findings in other industrialized nations.15–17
The proportion of patients admitted to ICUs with a diagnosis of sepsis who will progress to developing CCI is difficult to determine. Investigations suggest that, each year, more than 100,000 patients in the United States require prolonged mechanical ventilation.,18,19 Older studies estimated the annual cost of CCI to exceed $20 billion. This burden has undoubtedly already increased and, in the absence of a change in healthcare policy, will become more pronounced.20
Determining the actual mortality from sepsis is equally difficult. Application of evidence-based guidelines for sepsis management indicates a dramatic decline in mortality.21 Indeed, recent data suggest that mortality may be reduced to <10%.22 With use of diagnostic and management guidelines for sepsis, patients rarely die of shock. Most develop CCI and expire when exogenous support is discontinued (Deutschman CS, unpublished data). The cause of death in almost all cases is unknown; although patients die with CCI, the actual cause of death remains unclear.
PATHOPHYSIOLOGY OF SEPSIS, MODS, AND CHRONIC CRITICAL ILLNESS
As vexing as understanding the pathophysiology of sepsis may be, the mechanisms underlying MODS and CCI are even more problematic.
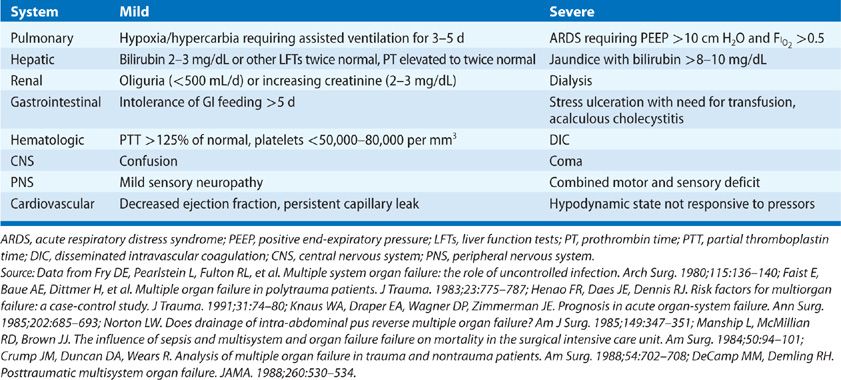
MODS was once thought to reflect the effects of a sustained activation of the immune system. However, the findings previously cited regarding early immunosuppression cast doubt on this hypothesis.7 Further, defining “dysfunction” in individual organs is problematic. Although many criteria have been used, none are universally accepted. Some generally used criteria for organ dysfunction are detailed in (Table 142-2).23 However, these criteria reflect the gaps in our understanding of MODS.
Recent studies suggest that early organ dysfunction differs from the organ failure that becomes evident later in the time course of sepsis. The early changes may represent an adaptive response aimed at preventing further damage from a severe insult.24–26 The basis for this construct lies in the “mitochondrial hypothesis,” which is detailed below. Development of CCI appears to herald the highly lethal state once attributed to MODS. The major factor limiting treatment of MODS is lack of a clear understanding of the underlying pathophysiologic defect. In fact, if not considered carefully, the changes associated with sepsis/MODS may simply resemble an extension of those observed after uncomplicated stress. For example, patients recovering from major surgery undergo increases in metabolic rate, oxygen consumption (![]() O2), and carbon dioxide production, in addition to glucose intolerance and hyperglycemia. The vasculature is dilated, and CO increases to promote oxygen transport. In addition, lactate production may increase, reflecting the overall increase in metabolism rather than tissue hypoxia.
O2), and carbon dioxide production, in addition to glucose intolerance and hyperglycemia. The vasculature is dilated, and CO increases to promote oxygen transport. In addition, lactate production may increase, reflecting the overall increase in metabolism rather than tissue hypoxia.
Typically, the septic patient presents with high fever, shock, and respiratory failure. This clinical picture resulted in the prevailing theory that sepsis reflects uncontrolled inflammation. The hypothesis was consistent with demonstration of increased concentrations of TNF, IL-1, IL-6 and, later in the time course, HMGB1 in sepsis.27 However, a myriad of trials addressing anti-inflammatory approaches to treatment have not demonstrated reduced survival rates.28,29 Two observations that may contribute to the negative findings have been offered. First, sepsis is characterized by both proinflammatory and anti-inflammatory responses. Second, as previously noted, immunosuppression in sepsis has been reported.6,7 Indeed, in a study of patients whose deaths were attributed to sepsis,30 80% had unresolved foci of infection. Hence, a strategy employing immune enhancement, rather than anti-inflammation, may be warranted. Further complicating an understanding of pathogenesis is our inability to define the underlying physiologic basis by which sepsis ultimately leads to organ failure or death. Studies6 have shown that there is very little cell death in most major organs in sepsis, and that histologic damage does not correlate with the degree of organ dysfunction seen prior to death. The exception to this observation lies in the immune system and in the gastrointestinal tract.31
Recent studies also suggest that organ dysfunction develops very early after the diagnosis of septic shock or severe sepsis is made. This finding is not surprising, given that both severe sepsis and septic shock are defined in terms of organ dysfunction.
The 2001 SCCM/ESICM definitions of sepsis were explicit in delineating specific clinical criteria for dysfunction in specific organs.5 In general, these abnormalities are not manifest for several days. However, when biochemical or histologic criteria are applied, organ dysfunction may be noted once the initial resuscitation is complete. The most common clinical abnormality is respiratory failure requiring mechanical ventilation. In some instances, the need for exogenous support of other organ systems (e.g., dialysis) may develop in short order, while in others deterioration may be slower. In either case, the patient progresses to development of CCI. These patients account for 5% to 10% of the ICU population. Most often, affected patients require prolonged mechanical ventilation, but they also may need continuous hemodialysis, cardiovascular support with vasopressors, prolonged exogenous metabolic/nutritional support, or complex wound management. One key characteristic of CCI is evident in ICU survivors32: These individuals suffer from prolonged physical symptoms, such as respiratory insufficiency and muscle weakness. Recovering patients are also often left with long-term neuropsychiatric impairment and reduced quality of life. Indeed, data suggest significant cognitive impairment in this cohort.33,34 Many suffer from posttraumatic stress disorder (PTSD).35 Perhaps, most alarming, data from the United States, Canada, and Australia demonstrate that ICU survivors are two to five times more likely to die compared with age- and sex-matched population controls.36–38 Three-year mortality in sepsis survivors is 70%; 5-year survival is 75%.14
A better understanding of these issues has emerged from the Surviving Sepsis Campaign (SSC),39 a program designed to enhance use of evidence-based approaches to sepsis management. Application of some or all of the elements outlined in SSC has dramatically reduced mortality in this early phase of the disorder.22 Future examination of the effects of the SSC and correlation with an emerging understanding of pathophysiology may help clarify matters.
UNDERLYING MECHANISMS: NEWER HYPOTHESES
As a result of advances in our understanding of the pathophysiology of sepsis, a number of hypotheses have been abandoned. In particular, those that proposed that sepsis reflected only the effects of excessive, uncontrolled inflammation are difficult to reconcile with recent data demonstrating early immunosuppression. Similarly, it has been difficult to demonstrate translocation of bacteria from the gut or the occurrence of “two hits,” that is, a scenario in which a second insult precipitates organ dysfunction. Several contemporary alternative hypotheses have emerged.
 THE MICROCIRCULATORY HYPOTHESIS
THE MICROCIRCULATORY HYPOTHESIS
Characteristic features of CCI are a defect in ![]() O2 and an increase in the production of lactate. Two theories have been proposed to account for these observations. The first, termed the microcirculatory hypothesis, posits that aberrant oxygen extraction results from a failure of cells or organs to receive adequate levels of oxygen or some important nutrient or substrate.40 Low blood flow, as is likely to occur in hypotension or shock, contributes to cellular dysfunction. The release of vasoactive mediators and vascular congestion secondary to microthrombi and leukocytes are also thought to play important roles.
O2 and an increase in the production of lactate. Two theories have been proposed to account for these observations. The first, termed the microcirculatory hypothesis, posits that aberrant oxygen extraction results from a failure of cells or organs to receive adequate levels of oxygen or some important nutrient or substrate.40 Low blood flow, as is likely to occur in hypotension or shock, contributes to cellular dysfunction. The release of vasoactive mediators and vascular congestion secondary to microthrombi and leukocytes are also thought to play important roles.
Reperfusion of ischemic tissue may be as important a determinant of tissue injury as is decreased flow itself.41,42 In particular, the generation of oxygen-free radicals and peroxidation of membrane lipids following reperfusion may contribute to tissue injury. Sources of free radicals include the conversion of molecular oxygen to superoxide by xanthine oxidase, activated leukocytes, mitochondria, and prostaglandin synthase.
Circulatory shock, microvascular compromise, and free radical generation likely directly affect the endothelium. Endothelial cells actively generate free radicals, provide a point of attachment for leukocytes, and may be exquisitely sensitive to hypoxia. In addition, they produce and respond to vasoactive mediators.43 As a result of these interactions, the microvascular hypothesis may be viewed as an extension of the cytokine hypothesis.44 Indeed, cytokines activate endothelial cells to elaborate other vasoactive substances and to express surface proteins that promote leukocyte adhesion. Further, endothelial cells are important participants in the formation of microthrombi.
The microvascular hypothesis is supported by autopsy data documenting the presence of microvascular injury and microthrombi containing platelets, neutrophils, and fibrin in patients dying with CCI. The use of side-stream, dark-field videomicroscopy has demonstrated microcirculatory failure in septic patients, while increased microcirculatory flow during resuscitation is associated with reduced organ failure at 24 hours, even in the absence of substantial changes in hemodynamics.45 Conversely, antibodies to CD18, which block leukocyte adhesion and are protective in some forms of ischemia–reperfusion injury,23,46 do not protect against liver injury or leukocyte adherence in experimental sepsis.46,47
While microcirculatory failure is present in sepsis and may contribute to the development of MODS and CCI, it cannot explain many of the clinical and pathologic features of these syndromes.
 THE MITOCHONDRIAL HYPOTHESIS
THE MITOCHONDRIAL HYPOTHESIS
An alternative explanation of the sepsis-induced defect in oxygen extraction is based on studies addressing mitochondrial dysfunction. The central tenet of this process, termed “cytopathic hypoxia,” is a defect in oxidative phosphorylation. In support of this concept is demonstration of impairment in several of the five complexes that comprise the mitochondrial respiratory chain in humans during experimental sepsis. A decrease in the so-called “Complex I” (NADH: ubiquinone oxidoreductase) activity is observed in skeletal muscle in patients with sepsis and in liver and muscle in septic rats.48,49 In addition, sepsis decreased Complexes II–III (succinate dehydrogenase and cytochrome bc1, respectively) activity has been reported in the hearts of septic rats,50 while attenuated Complex IV (cytochrome c oxidase) activity has been reported in cardiac muscle and liver in septic rats.25,51,52 Others have suggested that Complex IV functions as a sensor of dysoxia.53–58 Thus, inhibition of this enzyme complex in response to an insult limits oxidative phosphorylation and protects mitochondria.
 THE “FAILED COMMUNICATION” HYPOTHESIS
THE “FAILED COMMUNICATION” HYPOTHESIS
In the 1980s, investigators59,60
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


